Overdose
There is no specific antidote for overdose with Sutent and treatment of overdose should consist of general supportive measures. If indicated, elimination of unabsorbed active substance may be achieved by emesis or gastric lavage. Cases of overdose have been reported; some cases were associated with adverse reactions consistent with the known safety profile of sunitinib.
Shelf life
3 years.
Incompatibilities
Not applicable.
List of excipients
12.5 mg hard capsules
Capsule content
Mannitol (E421)
Croscarmellose sodium
Povidone (K-25)
Magnesium stearate
Capsule shell
Gelatin
Red iron oxide (E172)
Titanium dioxide (E171)
Printing ink
Shellac
Propylene glycol
Sodium hydroxide
Povidone
Titanium dioxide (E171)
25 mg hard capsules
Capsule content
Mannitol (E421)
Croscarmellose sodium
Povidone (K-25)
Magnesium stearate
Capsule shell
Gelatin
Red iron oxide (E172)
Titanium dioxide (E171)
Yellow iron oxide (E172)
Black iron oxide (E172)
Printing ink
Shellac
Propylene glycol
Sodium hydroxide
Povidone
Titanium dioxide (E171)
37.5 mg hard capsules
Capsule content
Mannitol (E421)
Croscarmellose sodium
Povidone (K-25)
Magnesium stearate
Capsule shell
Gelatin
Titanium dioxide (E171)
Yellow iron oxide (E172)
Printing ink
Shellac
Propylene glycol
Potassium hydroxide
Black iron oxide (E172)
50 mg hard capsules
Capsule content
Mannitol (E421)
Croscarmellose sodium
Povidone (K-25)
Magnesium stearate
Capsule shell
Gelatin
Titanium dioxide (E171)
Yellow iron oxide (E172)
Red iron oxide (E172)
Black iron oxide (E172)
Printing ink
Shellac
Propylene glycol
Sodium hydroxide
Povidone
Titanium dioxide (E171)
Pharmaceutical form
Hard capsule.
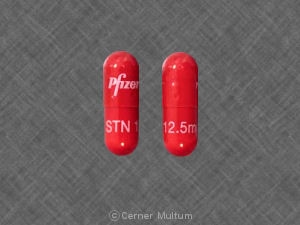
Sutent 12.5 mg hard capsules
Gelatin capsules with orange cap and orange body, printed with white ink “Pfizer†on the cap, “STN 12.5 mg†on the body, and containing yellow to orange granules.
Sutent 25 mg hard capsules
Gelatin capsules with caramel cap and orange body, printed with white ink “Pfizer†on the cap, “STN 25 mg†on the body, and containing yellow to orange granules.
Sutent 37.5 mg hard capsules
Gelatin capsules with yellow cap and yellow body, printed with black ink “Pfizer†on the cap, “STN 37.5 mg†on the body, and containing yellow to orange granules.
Sutent 50 mg hard capsules
Gelatin capsules with caramel cap and caramel body, printed with white ink “Pfizer†on the cap, “STN 50 mg†on the body, and containing yellow to orange granules.
Undesirable effects
Summary of the safety profile
The most serious adverse reactions associated with sunitinib, some fatal, are renal failure, heart failure, pulmonary embolism, gastrointestinal perforation, and haemorrhages (e.g., respiratory tract, gastrointestinal, tumour, urinary tract, and brain haemorrhages). The most common adverse reactions of any grade (experienced by patients in RCC, GIST, and pNET registrational trials) included decreased appetite, taste disturbance, hypertension, fatigue, gastrointestinal disorders (i.e. diarrhoea, nausea, stomatitis, dyspepsia, and vomiting), skin discolouration, and palmar-plantar erythrodysaesthesia syndrome. These symptoms may diminish as treatment continues. Hypothyroidism may develop during treatment. Haematological disorders (e.g., neutropenia, thrombocytopenia, and anaemia) are amongst the most common adverse drug reactions.
8 below that were considered possibly related to sunitinib included multi-system organ failure, disseminated intravascular coagulation, peritoneal haemorrhage, adrenal insufficiency, pneumothorax, shock, and sudden death.
Tabulated list of adverse reactions
Adverse reactions that were reported in GIST, MRCC, and pNET patients in a pooled dataset of 7,115 patients are listed below, by system organ class, frequency and grade of severity (NCI-CTCAE). Post- marketing adverse reactions identified in clinical studies are also included. Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Frequencies are defined as: very common (>1/10), common (>1/100 to <1/10), uncommon (>1/1,000 to <1/100), rare (>1/10,000 to <1/1,000), very rare (<1/10,000), not known (cannot be estimated from the available data).
Table 1. Adverse reactions reported in clinical trials
|
System organ class
|
Very common
|
Common
|
Uncommon
|
Rare
|
|
Infections and infestations
|
|
Viral infectionsa
Respiratory infectionsb,*
Abscessc,*
Fungal infectionsd
Urinary tract infection
Skin infectionse
Sepsisf,*
|
Necrotising fasciitis*
Bacterial infectionsg
|
|
|
Blood and lymphatic system disorders
|
Neutropoenia
Thrombocytopoenia
Anaemia
Leukopoenia
|
Lymphopoenia
|
Pancytopenia
|
Thrombotic microangiopathyh,*
|
|
Immune system disorders
|
|
|
Hypersensitivity
|
Angioedema
|
|
Endocrine disorders
|
Hypothyroidism
|
|
Hyperthyroidism
|
Thyroiditis
|
|
Metabolism and nutrition disorders
|
Decreased appetitei
|
Dehydration
Hypoglycaemia
|
|
Tumour lysis syndrome*
|
|
Psychiatric disorders
|
Insomnia
|
Depression
|
|
|
|
Nervous system disorders
|
Dizziness
Headache
Taste disturbancej
|
Neuropathy peripheral
Paraesthesia
Hypoaesthesia
Hyperaesthesia
|
Cerebral haemorrhage*
Cerebrovascular accident*
Transient ischaemic attack
|
Posterior reversible encephalopathy syndrome*
|
|
Eye disorders
|
|
Periorbital oedema
Eyelid oedema
Lacrimation increased
|
|
|
|
Cardiac disorders
|
|
Myocardial ischemiak,*
Ejection fraction decreasedl
|
Cardiac failure congestive
Myocardial infarctionm,*
Cardiac failure*
Cardiomyopathy*
Pericardial effusion
Electrocardiogram QT prolonged
|
Left ventricular failure*
Torsade de pointes
|
|
Vascular disorders
|
Hypertension
|
Deep vein thrombosis
Hot flush
Flushing
|
Tumour haemorrhage*
|
|
|
Respiratory, thoracic and mediastinal disorders
|
Dyspnoea
Epistaxis
Cough
|
Pulmonary embolism*
Pleural effusion*
Haemoptysis
Dyspnoea exertional
Oropharyngeal painn
Nasal congestion
Nasal dryness
|
Pulmonary haemorrhage*
Respiratory failure*
|
|
|
Gastrointestinal disorders
|
Stomatitiso
Abdominal painp
Vomiting
Diarrhoea
Dyspepsia
Nausea
Constipation
|
Gastro-oesophageal reflux disease
Dysphagia
Gastrointestinal haemorrhage*
Oesophagitis*
Abdominal distension
Abdominal discomfort
Rectal haemorrhage
Gingival bleeding
Mouth ulceration
Proctalgia
Cheilitis
Haemorrhoids
Glossodynia
Oral pain
Dry mouth
Flatulence
Oral discomfort
Eructation
|
Gastrointestinal perforationq,*
Pancreatitis
Anal fistula
|
|
|
Hepatobiliary disorders
|
|
|
Hepatic failure*
Cholecystitisr,*
Hepatic function abnormal
|
Hepatitis
|
|
Skin and subcutaneous tissue disorders
|
Skin discolourations
Palmar-plantar erythrodysaesthesia syndrome
Rasht
Hair colour changes
Dry skin
|
Skin exfoliation
Skin reactionu
Eczema
Blister
Erythema
Alopecia
Acne
Pruritus
Skin hyperpigmentation
Skin lesion
Hyperkeratosis
Dermatitis
Nail disorderv
|
|
Erythema multiforme*
Stevens-Johnson syndrome*
Pyoderma gangrenosum
Toxic epidermal necrolysis*
|
|
Musculoskeletal and connective tissue disorders
|
Pain in extremity
Arthralgia
Back pain
|
Musculoskeletal pain
Muscle spasms
Myalgia
Muscular weakness
|
Osteonecrosis of the jaw
Fistula*
|
Rhabdomyolysis*
Myopathy
|
|
Renal and urinary disorders
|
|
Renal failure*
Renal failure acute*
Chromaturia
Proteinuria
|
Haemorrhage urinary tract
|
Nephrotic syndrome
|
|
General disorders and administration site conditions
|
Mucosal inflammation
Fatiguew
Oedemax
Pyrexia
|
Chest pain
Pain
Influenza like illness
Chills
|
Impaired healing
|
|
|
Investigations
|
|
Weight decreased
White blood cell count decreased
Lipase increased
Platelet count decreased
Haemoglobin decreased
Amylase increasedy
Aspartate aminotransferase increased
Alanine aminotransferase increased
Blood creatinine increased
Blood pressure increased
Blood uric acid increased
|
Blood creatine phosphokinase increased
Blood thyroid stimulating hormone increased
|
|
* Including fatal events
The following terms have been combined:
a Nasopharyngitis and oral herpes.
b Bronchitis, lower respiratory tract infection, pneumonia, and respiratory tract infection.
c Abscess, abscess limb, anal abscess, gingival abscess, liver abscess, pancreatic abscess, perineal abscess, perirectal abscess, rectal abscess, subcutaneous abscess, and tooth abscess.
d Oesophageal candidiasis and oral candidiasis.
e Cellulitis and skin infection.
f Sepsis and sepsis shock.
g Abdominal abscess, abdominal sepsis, diverticulitis, and osteomyelitis.
h Thrombotic microangiopathy, thrombotic thrombocytopenic purpura, and haemolytic uraemic syndrome.
i Decreased appetite and anorexia
j Dysgeusia, ageusia, and taste disturbance.
k Acute coronary syndrome, angina pectoris, angina unstable, coronary artery occlusion, and myocardial ischaemia.
l Ejection fraction decreased/abnormal.
m Acute myocardial infarction, myocardial infarction, and silent myocardial infarction.
n Oropharyngeal and pharyngolaryngeal pain.
o Stomatitis and aphtous stomatitis.
p Abdominal pain, abdominal pain lower, and abdominal pain upper.
q Gastrointestinal perforation and intestinal perforation.
r Cholecystitis and acalculous cholecystitis.
s Yellow skin, skin discolouration, and pigmentation disorder.
t Dermatitis psoriasiform, exfoliative rash, rash, rash erythematous, rash follicular, rash generalised, rash macular, rash maculo-papular, rash papular, and rash pruritic.
u Skin reaction and skin disorder.
v Nail disorder and discolouration.
w Fatigue and asthenia.
x Face oedema, oedema, and oedema peripheral.
y Amylase and amylase increased.
Description of selected adverse reactions
Infections and infestations
Cases of serious infection (with or without neutropenia), including cases with fatal outcome, have been reported.).
Blood and lymphatic system disorders
Decreased absolute neutrophil counts of Grade 3 and 4 severities, respectively, were reported in 10% and 1.7% of patients on the Phase 3 GIST study, in 16% and 1.6% of patients on the Phase 3 MRCC study, and in 13% and 2.4% of patients on the Phase 3 pNET study. Decreased platelet counts of Grade 3 and 4 severities, respectively, were reported in 3.7% and 0.4% of patients on the Phase 3 GIST study, in 8.2% and 1.1% of patients on the Phase 3 MRCC study, and in 3.7% and 1.2% of patients on the Phase 3 pNET study.
Bleeding events were reported in 18% of patients receiving sunitinib in a Phase 3 GIST study vs 17% of patients receiving placebo. In patients receiving sunitinib for treatment-naïve MRCC, 39% had bleeding events vs 11% of patients receiving interferon-α (IFN-α). Seventeen (4.5%) patients on sunitinib versus 5 (1.7%) patients on IFN-α experienced Grade 3 or greater bleeding events. Of patients receiving sunitinib for cytokine-refractory MRCC, 26% experienced bleeding. Bleeding events, excluding epistaxis, were reported in 21.7% of patients receiving sunitinib in the Phase 3 pNET study compared to 9.85% of patients receiving placebo
In clinical trials, tumour haemorrhage was reported in approximately 2% of patients with GIST.
Immune system disorders
Hypersensitivity reactions, including angioedema, have been reported.
Endocrine disorders
Hypothyroidism was reported as an adverse reaction in 7 patients (4%) receiving sunitinib across the 2 cytokine-refractory MRCC studies; in 61 patients (16%) on sunitinib and 3 patients (< 1%) in the IFN-α arm in the treatment-naïve MRCC study.
Additionally, thyroid-stimulating hormone (TSH) elevations were reported in 4 cytokine-refractory MRCC patients (2%). Overall, 7% of the MRCC population had either clinical or laboratory evidence of treatment-emergent hypothyroidism. Acquired hypothyroidism was noted in 6.2% of GIST patients on sunitinib versus 1% on placebo. In the Phase 3 pNET study hypothyroidism was reported in 6 patients (7.2%) receiving sunitinib and in 1 patient (1.2%) on placebo.
Thyroid function was monitored prospectively in 2 studies in patients with breast cancer; Sutent is not approved for use in breast cancer. In 1 study, hypothyroidism was reported in 15 (13.6%) patients on sunitinib and 3 (2.9%) patients on standard of care. Blood TSH increase was reported in 1 (0.9%) patient on sunitinib and no patients on standard of care. Hyperthyroidism was reported in no sunitinib-treated patients and 1 (1.0%) patient receiving standard of care. In the other study hypothyroidism was reported in a total of 31 (13%) patients on sunitinib and 2 (0.8%) patients on capecitabine. Blood TSH increase was reported in 12 (5.0%) patients on sunitinib and no patients on capecitabine. Hyperthyroidism was reported in 4 (1.7%) patients on sunitinib and no patients on capecitabine. Blood TSH decrease was reported in 3 (1.3%) patients on sunitinib and no patients on capecitabine. T4 increase was reported in 2 (0.8%) patients on sunitinib and 1 (0.4%) patient on capecitabine. T3 increase was reported in 1 (0.8%) patient on sunitinib and no patients on capecitabine. All thyroid-related events reported were Grade 1-2.
Metabolism and nutrition disorders
A higher incidence rate of hypoglycaemia events was reported in patients with pNET in comparison to MRCC and GIST. Nevertheless, most of these adverse events observed in clinical studies were not considered related to study treatment.
Nervous system disorders
In clinical studies of sunitinib and from postmarketing surveillance, there have been few reports (< 1%), some fatal, of subjects presenting with seizures and radiological evidence of RPLS. Seizures have been observed in patients with or without radiological evidence of brain metastases.
Cardiac disorders
In clinical trials, decreases in left ventricular ejection fraction (LVEF) of > 20% and below the lower limit of normal were reported in approximately 2% of sunitinib-treated GIST patients, 4% of cytokine-refractory MRCC patients, and 2% of placebo-treated GIST patients. These LVEF declines do not appear to have been progressive and often improved as treatment continued. In the treatment-naïve MRCC study, 27% of patients on sunitinib and 15% of patients on IFN-α had an LVEF value below the lower limit of normal. Two patients (< 1%) who received sunitinib were diagnosed with CHF.
In GIST patients 'cardiac failure', 'cardiac failure congestive', or 'left ventricular failure' were reported in 1.2% of patients treated with sunitinib and 1% of patients treated with placebo. In the pivotal Phase 3 GIST study (N = 312), treatment-related fatal cardiac reactions were reported in 1% of patients on each arm of the study (i.e. sunitinib and placebo arms). In a Phase 2 study in cytokine-refractory MRCC patients, 0.9% of patients experienced treatment-related fatal myocardial infarction and in the Phase 3 study in treatment-naïve MRCC patients, 0.6% of patients on the IFN-α arm and 0% of patients on the sunitinib arm experienced fatal cardiac events. In the Phase 3 pNET study, 1 (1%) patient who received sunitinib had treatment-related fatal cardiac failure.
Hypertension
Hypertension was a very common adverse reaction reported in clinical trials. The dose of sunitinib was reduced or its administration temporarily suspended in approximately 2.7% of the patients who experienced hypertension. Sunitinib was not permanently discontinued in any of these patients. Severe hypertension (> 200 mmHg systolic or 110 mmHg diastolic) was reported in 4.7% of patients with solid tumours. Hypertension was reported in approximately 33.9% of patients receiving sunitinib for treatment-naïve MRCC compared to 3.6% of patients receiving IFN-α. Severe hypertension was reported in 12% of treatment-naïve patients on sunitinib and < 1% of patients on IFN-α. Hypertension was reported in 26.5% of patients receiving sunitinib in a Phase 3 pNET study, compared to 4.9% of patients receiving placebo. Severe hypertension was reported in 10% of pNET patients on sunitinib and 3% of patients on placebo.
Venous thromboembolic events
Treatment-related venous thromboembolic events were reported in approximately 1.0% of patients with solid tumours who received sunitinib on clinical trials, including GIST and RCC.
Seven patients (3%) on sunitinib and none on placebo in a Phase 3 GIST study experienced venous thromboembolic events; 5 of the 7 were Grade 3 deep venous thrombosis (DVT) and 2 were Grade 1 or 2. Four of these 7 GIST patients discontinued treatment following first observation of DVT.
Thirteen patients (3%) receiving sunitinib in the Phase 3 treatment-naïve MRCC study and 4 patients (2%) on the 2 cytokine-refractory MRCC studies had venous thromboembolic events reported. Nine of these patients had pulmonary embolisms; 1 was Grade 2 and 8 were Grade 4. Eight of these patients had DVT; 1 with Grade 1, 2 with Grade 2, 4 with Grade 3, and 1 with Grade 4. One patient with pulmonary embolism in the cytokine-refractory MRCC study experienced dose interruption.
In treatment-naïve MRCC patients receiving IFN-α, 6 (2%) venous thromboembolic events were reported; 1 patient (< 1%) experienced a Grade 3 DVT and 5 patients (1%) had pulmonary embolisms, all with Grade 4.
Venous thromboembolic events were reported for 1 (1.2%) patient in the sunitinib arm and 5 (6.1%) patients in the placebo arm in the Phase 3 pNET study. Two of these patients on placebo had DVT, 1 with Grade 2 and 1 with Grade 3.
No cases with fatal outcome were reported in GIST, MRCC, and pNET registrational studies. Cases with fatal outcome have been observed in the postmarketing surveillance.
Cases of pulmonary embolism were observed in approximately 3.1% of patients with GIST and in approximately 1.2% of patients with MRCC, who received sunitinib in Phase 3 studies. No pulmonary embolism was reported for patients with pNET who received sunitinib in the Phase 3 study. Rare cases with fatal outcome have been observed in the postmarketing surveillance.
Patients who presented with pulmonary embolism within the previous 12 months were excluded from sunitinib clinical studies.
In patients who received sunitinib in Phase 3 registrational studies, pulmonary events (i.e. dyspnoea, pleural effusion, pulmonary embolism, or pulmonary oedema) were reported in approximately 17.8% of patients with GIST, in approximately 26.7% of patients with MRCC and in 12% of patients with pNET.
Approximately 22.2% of patients with solid tumours, including GIST and MRCC, who received sunitinib in clinical trials experienced pulmonary events.
Gastrointestinal disorders
Pancreatitis has been observed uncommonly (< 1%) in patients receiving sunitinib for GIST or MRCC. No treatment-related pancreatitis was reported in the Phase 3 pNET study.
Fatal gastrointestinal bleeding was reported in 0.98% of patients receiving placebo in the GIST Phase 3 study.
Hepatobiliary disorders
Hepatic dysfunction has been reported and may include Liver Function Test abnormalities, hepatitis, or liver failure.
Skin and subcutaneous tissue disorders
Musculoskeletal and connective tissue disorders
Cases of myopathy and/or rhabdomyolysis, some with acute renal failure, have been reported. Patients with signs or symptoms of muscle toxicity should be managed as per standard medical practice.
Cases of fistula formation, sometimes associated with tumour necrosis and regression, in some cases with fatal outcomes, have been reported.
Investigations
Data from non clinical (in vitro and in vivo) studies, at doses higher than the recommended human dose, indicated that sunitinib has the potential to inhibit the cardiac action potential repolarisation process (e.g., prolongation of QT interval).
Increases in the QTc interval to over 500 msec were reported in 0.5%, and changes from baseline in excess of 60 msec were reported in 1.1% of the 450 solid tumour patients; both of these parameters are recognised as potentially significant changes. At approximately twice therapeutic concentrations, sunitinib has been shown to prolong the QTcF interval (Fridericia corrected QT interval).
QTc interval prolongation was investigated in a trial in 24 patients, ages 20-87 years, with advanced malignancies. The results of this study demonstrated that sunitinib had an effect on QTc interval (defined as a mean placebo-adjusted change of > 10 msec with a 90% confidence interval [CI] upper limit > 15 msec) at therapeutic concentration (Day 3) using the within-day baseline correction method, and at greater than therapeutic concentration (Day 9) using both baseline correction methods. No patients had a QTc interval > 500 msec. Although an effect on QTcF interval was observed on Day 3 at 24 hours postdose (i.e., at therapeutic plasma concentration expected after the recommended starting dose of 50 mg) with the within-day baseline correction method, the clinical significance of this finding is unclear.
Using comprehensive serial ECG assessments at times corresponding to either therapeutic or greater than therapeutic exposures, none of the patients in the evaluable or intent-to-treat (ITT) populations were observed to develop QTc interval prolongation considered as “severe†(i.e. equal to or greater than Grade 3 by Common Terminology Criteria for Adverse Events [CTCAE] version 3.0).
At therapeutic plasma concentrations, the maximum QTcF interval (Frederica's correction) mean change from baseline was 9 msec (90% CI: 15.1 msec). At approximately twice therapeutic concentrations, the maximum QTcF interval change from baseline was 15.4 msec (90% CI: 22.4 msec). Moxifloxacin (400 mg) used as a positive control showed a 5.6 msec maximum mean QTcF interval change from baseline. No subjects experienced an effect on the QTc interval greater than Grade 2 (CTCAE version 3.0).
Long-term safety in MRCC
The long-term safety of sunitinib in patients with MRCC was analysed across 9 completed clinical studies conducted in the first-line, bevacizumab-refractory, and cytokine-refractory treatment settings in 5,739 patients, of whom 807 (14%) were treated for > 2 years up to 6 years. In the 807 patients who received long-term sunitinib treatment, most treatment-related adverse events (TRAEs) occurred initially in the first 6 months-1 year and then were stable or decreased in frequency over time, with the exception of hypothyroidism, which gradually increased over time, with new cases occurring over the 6 year period. Prolonged treatment with sunitinib did not appear to be associated with new types of TRAEs.
Paediatric population
A Phase 1 dose-escalation study of oral sunitinib was conducted in 35 paediatric and young adult patients (aged 2-21) with refractory solid tumours, the majority of whom had a primary diagnosis of brain tumour. All study participants experienced adverse drug reactions and in those patients with previous exposure to anthracyclines or cardiac radiation most of these were severe (toxicity grade > 3) and included cardiac toxicity. The risk of cardiac adverse drug reactions appears higher in paediatric patients with previous exposure to cardiac radiation and anthracycline, compared to those paediatric patients without previous exposure. No maximum tolerated dose of sunitinib has been identified for this patient population due to dose-limiting toxicities. In paediatric patients without previous exposure to anthracyclines or cardiac radiation, the most common adverse reactions were gastrointestinal (GI) toxicity, neutropenia, fatigue, and ALT elevation.
Based on a population pharmacokinetics (PK) and pharmacokinetic pharmacodynamic (PK/PD) analysis, sunitinib at doses of 25 mg/m2/day on Schedule 4/2 in paediatric patients (ages: 6-11 and 12-17 years) with GIST is predicted to provide comparable sunitinib plasma exposures, and subsequently safety and efficacy profiles, to those in adult patients with GIST treated at 50 mg/day on Schedule 4/2.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product.
United Kingdom
Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at www.mhra.gov.uk/yellowcard or search for MHRA Yellow Card in the Google Play or Apple App Store.
Ireland
Healthcare professionals are asked to report any suspected adverse reactions via HPRA Pharmacovigilance, Earlsfort Terrace, IRL - Dublin 2, Tel: +353 1 6764971, Fax: +353 1 6762517, Website: www.hpra.ie E-mail: [email protected]
Preclinical safety data
In rat and monkey repeated-dose toxicity studies up to 9-months duration, the primary target organ effects were identified in the gastrointestinal tract (emesis and diarrhoea in monkeys); adrenal gland (cortical congestion and/or haemorrhage in rats and monkeys, with necrosis followed by fibrosis in rats); haemolymphopoietic system (bone morrow hypocellularity and lymphoid depletion of thymus, spleen, and lymph node); exocrine pancreas (acinar cell degranulation with single cell necrosis); salivary gland (acinar hypertrophy); bone joint (growth plate thickening); uterus (atrophy); and ovaries (decreased follicular development). All findings occurred at clinically relevant sunitinib plasma exposure levels. Additional effects observed in other studies included: QTc interval prolongation, LVEF reduction and testicular tubular atrophy, increased mesangial cells in kidney, haemorrhage in gastro-intestinal tract and oral mucosa, and hypertrophy of anterior pituitary cells. Changes in the uterus (endometrial atrophy) and bone growth plate (physeal thickening or dysplasia of cartilage) are thought to be related to the pharmacological action of sunitinib. Most of these findings were reversible after 2 to 6 weeks without treatment.
Genotoxicity
The genotoxic potential of sunitinib was assessed in vitro and in vivo. Sunitinib was not mutagenic in bacteria using metabolic activation provided by rat liver. Sunitinib did not induce structural chromosome aberrations in human peripheral blood lymphocyte cells in vitro. Polyploidy (numerical chromosome aberrations) was observed in human peripheral blood lymphocytes in vitro, both in the presence and absence of metabolic activation. Sunitinib was not clastogenic in rat bone marrow in vivo. The major active metabolite was not evaluated for genotoxic potential.
Carcinogenicity
In a 1-month, oral gavage dose-range finding study (0, 10, 25, 75, or 200 mg/kg/day) with continuous daily dosing in rasH2 transgenic mice, carcinoma and hyperplasia of Brunner's glands of the duodenum were observed at the highest dose (200 mg/kg/day) tested.
A 6-month, oral gavage carcinogenicity study (0, 8, 25, 75 [reduced to 50] mg/kg/day), with daily dosing was conducted in rasH2 transgenic mice. Gastroduodenal carcinomas, an increased incidence of background haemangiosarcomas, and/or gastric mucosal hyperplasia were observed at doses of > 25 mg/kg/day following 1- or 6-months duration (> 7.3 times the AUC in patients administered the recommended daily dose [RDD]).
In a 2-year rat carcinogenicity study (0, 0.33, 1, or 3 mg/kg/day), administration of sunitinib in 28-day cycles followed by 7-day dose-free periods resulted in increases in the incidence of phaeochromocytomas and hyperplasia in the adrenal medulla of male rats given 3 mg/kg/day following > 1 year of dosing (> 7.8 times the AUC in patients administered the RDD). Brunner's glands carcinoma occurred in the duodenum at > 1 mg/kg/day in females and at 3 mg/kg/day in males, and mucous cell hyperplasia was evident in the glandular stomach at 3 mg/kg/day in males, which occurred at > 0.9, 7.8, and 7.8 times the AUC in patients administered the RDD, respectively. The relevance to humans of the neoplastic findings observed in the mouse (rasH2 transgenic) and rat carcinogenicity studies with sunitinib treatment is unclear.
Reproductive and developmental toxicity
No effects on male or female fertility were observed in reproductive toxicity studies. However, in repeated-dose toxicity studies performed in rats and monkeys, effects on female fertility were observed in the form of follicular atresia, degeneration of corpora lutea, endometrial changes in the uterus, and decreased uterine and ovarian weights at clinically relevant systemic exposure levels. Effects on male fertility in rat were observed in the form of tubular atrophy in the testes, reduction of spermatozoa in epididymides, and colloid depletion in prostate and seminal vesicles at plasma exposure levels 25 times the systemic exposure in humans.
In rats, embryo-foetal mortality was evident as significant reductions in the number of live foetuses, increased numbers of resorptions, increased postimplantation loss, and total litter loss in 8 of 28 pregnant females at plasma exposure levels 5.5 times the systemic exposure in humans. In rabbits, reductions in gravid uterine weights and number of live foetuses were due to increases in the number of resorptions, increases in postimplantation loss and complete litter loss in 4 of 6 pregnant females at plasma exposure levels 3 times the systemic exposure in humans. Sunitinib treatment in rats during organogenesis resulted in developmental effects at > 5 mg/kg/day consisting of increased incidence of foetal skeletal malformations, predominantly characterised as retarded ossification of thoracic/lumbar vertebrae and occurred at plasma exposure levels 5.5 times the systemic exposure in humans. In rabbits, developmental effects consisted of increased incidence of cleft lip at plasma exposure levels approximately equal to that observed in clinic, and cleft lip and cleft palate at plasma exposure levels 2.7 times the systemic exposure in humans.
Sunitinib (0.3, 1.0, 3.0 mg/kg/day) was evaluated in a pre-and postnatal development study in pregnant rats. Maternal body weight gains were reduced during gestation and lactation at > 1 mg/kg/day but no maternal reproductive toxicity was observed up to 3 mg/kg/day (estimate exposure > 2.3 times the AUC in patients administered the RDD). Reduced offspring body weights were observed during the preweaning and postweaning periods at 3 mg/kg/day. No development toxicity was observed at 1 mg/kg/day (approximate exposure > 0.9 times the AUC in patients administered the RDD).
Therapeutic indications
Gastrointestinal stromal tumour (GIST)
Sutent is indicated for the treatment of unresectable and/or metastatic malignant gastrointestinal stromal tumour (GIST) in adults after failure of imatinib treatment due to resistance or intolerance.
Metastatic renal cell carcinoma (MRCC)
Sutent is indicated for the treatment of advanced/metastatic renal cell carcinoma (MRCC) in adults.
Pancreatic neuroendocrine tumours (pNET)
Sutent is indicated for the treatment of unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumours (pNET) with disease progression in adults.
Pharmacotherapeutic group
Antineoplastic agents, protein kinase inhibitors; ATC code: L01XE04
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors; ATC code: L01XE04
Mechanism of action
Sunitinib inhibits multiple RTKs that are implicated in tumour growth, neoangiogenesis, and metastatic progression of cancer. Sunitinib was identified as an inhibitor of platelet-derived growth factor receptors (PDGFRα and PDGFRβ), vascular endothelial growth factor receptors (VEGFR1, VEGFR2, and VEGFR3), stem cell factor receptor (KIT), Fms-like tyrosine kinase-3 (FLT3), colony stimulating factor receptor (CSF-1R), and the glial cell-line derived neurotrophic factor receptor (RET). The primary metabolite exhibits similar potency compared to sunitinib in biochemical and cellular assays.
Clinical efficacy and safety
The clinical safety and efficacy of sunitinib has been studied in the treatment of patients with GIST who were resistant to imatinib (i.e., those who experienced disease progression during or following treatment with imatinib) or intolerant to imatinib (i.e., those who experienced significant toxicity during treatment with imatinib that precluded further treatment), the treatment of patients with MRCC, and the treatment of patients with unresectable pNET.
Efficacy is based on time-to-tumour progression (TTP) and an increase in survival in GIST, on progression-free survival (PFS) and objective response rates (ORR) for treatment-naïve and cytokine-refractory MRCC respectively, and on PFS for pNET.
Gastrointestinal stromal tumours
An initial open-label, dose-escalation study was conducted in patients with GIST after failure of imatinib (median maximum daily dose 800 mg) due to resistance or intolerance. Ninety-seven patients were enrolled at various doses and schedules; 55 patients received 50 mg at the recommended treatment Schedule 4 weeks on /2 weeks off (“Schedule 4/2â€).
In this study, the median TTP was 34.0 weeks (95% CI: 22.0, 46.0).
A Phase 3, randomised, double-blind, placebo-controlled study of sunitinib was conducted in patients with GIST who were intolerant to, or had experienced disease progression during or following treatment with imatinib (median maximum daily dose 800 mg). In this study, 312 patients were randomised (2:1) to receive either 50 mg sunitinib or placebo, orally once daily on Schedule 4/2 until disease progression or withdrawal from the study for another reason (207 patients received sunitinib and 105 patients received placebo). The primary efficacy endpoint of the study was TTP, defined as the time from randomisation to first documentation of objective tumour progression. At the time of the prespecified interim analysis, the median TTP on sunitinib was 28.9 weeks (95% CI: 21.3, 34.1) as assessed by the investigator and 27.3 weeks (95% CI: 16.0, 32.1) as assessed by the independent review and was statistically significantly longer than the TTP on placebo of 5.1 weeks (95% CI: 4.4, 10.1) as assessed by the investigator and 6.4 weeks (95% CI: 4.4, 10.0) as assessed by the independent review. The difference in overall survival (OS) was statistically in favour of sunitinib [hazard ratio (HR): 0.491; (95% CI: 0.290, 0.831)]; the risk of death was 2 times higher in patients in the placebo arm compared to the sunitinib arm.
After the interim analysis of efficacy and safety, at the recommendation of the independent Data and Safety Monitoring Board (DSMB), the study was unblinded and patients on the placebo arm were offered open-label sunitinib treatment.
A total of 255 patients received sunitinib in the open-label treatment phase of the study, including 99 patients who were initially treated with placebo.
The analyses of primary and secondary endpoints in the open-label phase of the study reaffirmed the results obtained at the time of the interim analysis, as shown in Table 2:
Table 2. GIST summary of efficacy endpoints (ITT population)
|
|
Double-blind treatmenta
|
|
|
|
Median (95% CI)
|
Hazard ratio
|
Placebo cross-over group treatmentb
|
|
Endpoint
|
Sutent
|
Placebo
|
(95% CI)
|
p-value
|
|
Primary
|
|
|
|
|
|
|
TTP (weeks)
|
|
|
|
|
|
|
Interim
|
27.3 (16.0, 32.1)
|
6.4 (4.4, 10.0)
|
0.329 (0.233, 0.466)
|
< 0.001
|
-
|
|
Final
|
26.6 (16.0, 32.1)
|
6.4 (4.4, 10.0 )
|
0.339 (0.244, 0.472)
|
< 0.001
|
10.4 (4.3, 22.0)
|
|
Secondary
|
|
|
|
|
|
|
PFS (weeks)c
|
|
|
|
|
|
|
Interim
|
24.1 (11.1, 28.3)
|
6.0 (4.4, 9.9)
|
0.333 (0.238, 0.467)
|
< 0.001
|
-
|
|
Final
|
22.9 (10.9, 28.0)
|
6.0 (4.4, 9.7)
|
0.347 (0.253, 0.475)
|
< 0.001
|
-
|
|
ORR (%)d
|
|
|
|
|
|
|
Interim
|
6.8 (3.7, 11.1)
|
0 (-)
|
NA
|
0.006
|
-
|
|
Final
|
6.6 (3.8, 10.5)
|
0 (-)
|
NA
|
0.004
|
10.1 (5.0, 17.8)
|
|
OS (weeks)e
|
|
|
|
|
|
|
Interim
|
-
|
-
|
0.491 (0.290, 0.831)
|
0.007
|
-
|
|
Final
|
72.7 (61.3, 83.0)
|
64.9 (45.7, 96.0)
|
0.876 (0.679, 1.129)
|
0.306
|
-
|
|
Abbreviations: CI=confidence interval; ITT=intent-to-treat; NA=not applicable; ORR=objective response rate; OS=overall survival; PFS=progression-free survival; TTP=time-to-tumour progression.
a Results of double-blind treatment are from the ITT population and using central radiologist measurement, as appropriate.
b Efficacy results for the 99 subjects who crossed over from placebo to Sutent after unblinding. Baseline was reset at cross-over and efficacy analyses were based on investigators assessment.
c The interim PFS numbers have been updated based on a recalculation of the original data.
d Results for ORR are given as percent of subjects with confirmed response with the 95% CI.
e Median not achieved because the data were not yet mature.
|
Median OS in the ITT population was 72.7 weeks and 64.9 weeks (HR: 0.876; 95% CI: 0.679, 1.129; p = 0.306), in the sunitinib and placebo arms, respectively. In this analysis, the placebo arm included those patients randomised to placebo who subsequently received open-label sunitinib treatment.
Treatment-naïve metastatic renal cell carcinoma
A Phase 3, randomised, multi-centre, international study evaluating the efficacy and safety of sunitinib compared with IFN-α in treatment-naïve MRCC patients was conducted. Seven hundred and fifty patients were randomised 1:1 to the treatment arms; they received treatment with either sunitinib in repeated 6-week cycles, consisting of 4 weeks of 50 mg daily oral administration followed by 2 weeks of rest (Schedule 4/2), or IFN-α, administered as a subcutaneous injection of 3 million units (MU) the first week, 6 MU the second week, and 9 MU the third week and thereafter, on 3 nonconsecutive days each week.
The median duration of treatment was 11.1 months (range: 0.4-46.1) for sunitinib treatment and 4.1 months (range: 0.1-45.6) for IFN-α treatment. Treatment-related serious adverse events (TRSAEs) were reported in 23.7% of patients receiving sunitinib and in 6.9% of patients receiving IFN-α. However, the discontinuation rates due to adverse events were 20% for sunitinib and 23% for IFN-α. Dose interruptions occurred in 202 patients (54%) on sunitinib and 141 patients (39%) on IFN-α. Dose reductions occurred in 194 patients (52%) on sunitinib and 98 patients (27%) on IFN-α. Patients were treated until disease progression or withdrawal from the study. The primary efficacy endpoint was PFS. A planned interim analysis showed a statistically significant advantage for sunitinib over IFN-α, in this study, the median PFS for the sunitinib-treated group was 47.3 weeks, compared with 22.0 weeks for the IFN-α-treated group; the HR was 0.415 (95% CI: 0.320, 0.539; p-value < 0.001). Other endpoints included ORR, OS, and safety. Core radiology assessment was discontinued after the primary endpoint had been met. At the final analysis, the ORR as determined by the investigator's assessment was 46% (95% CI: 41%, 51%) for the sunitinib arm and 12.0% (95% CI: 9%, 16%) for the IFN-α arm (p<0.001).
Sunitinib treatment was associated with longer survival compared to IFN-α. The median OS was 114.6 weeks for the sunitinib arm (95% CI: 100.1, 142.9) and 94.9 weeks for the IFN-α arm (95% CI: 77.7, 117.0) with a hazard ratio of 0.821 (95% CI: 0.673, 1.001; p=0.0510 by unstratified log-rank).
The overall PFS and OS, observed in the ITT population, as determined by the core radiology laboratory assessment, are summarised in Table 3.
|
Table 3. Treatment-naïve mRCC summary of efficacy endpoints (ITT population)
|
|
|
Summary of progression-free survival
|
Sunitinib
(N = 375)
|
IFN-α
(N = 375)
|
|
Subject did not progress or die [n (%)]
|
161 (42.9)
|
176 (46.9)
|
|
Subject observed to have progressed or died [n (%)]
|
214 (57.1)
|
199 (53.1)
|
|
PFS (weeks)
|
|
Quartile (95% CI)
|
|
25%
|
22.7 (18.0, 34.0)
|
10.0 (7.3, 10.3)
|
|
50%
|
48.3 (46.4, 58.3)
|
22.1 (17.1, 24.0)
|
|
75%
|
84.3 (72.9, 95.1)
|
58.1 (45.6, 82.1)
|
|
Unstratified analysis
|
|
Hazard ratio (sunitinib versus IFN-α)
|
0.5268
|
|
95% CI for hazard ratio
|
(0.4316, 0.6430)
|
|
p-valuea
|
< 0.0001
|
|
Summary of overall survival
|
|
Subject not known to have died [n (%)]
|
185 (49.3)
|
175 (46.7)
|
|
Subject observed to have died [n (%)]
|
190 (50.7)
|
200 (53.3)
|
|
OS (weeks)
|
|
Quartile (95% CI)
|
|
25%
|
56.6 (48.7, 68.4)
|
41.7 (32.6, 51.6)
|
|
50%
|
114.6 (100.1, 142.9)
|
94.9 (77.7, 117.0)
|
|
75%
|
NA (NA, NA)
|
NA (NA, NA)
|
|
Unstratified analysis
|
|
Hazard ratio (sunitinib versus IFN-α)
|
0.8209
|
|
95% CI for hazard ratio
|
(0.6730, 1.0013)
|
|
p-valuea
|
0.0510
|
|
Abbreviations: CI=confidence interval; INF-α=interferon-alfa; ITT=intent-to-treat; N=number of patients; NA=not applicable; OS=overall survival; PFS=progression-free survival.
a From a 2-sided log-rank test.
|
Cytokine-refractory metastatic renal cell carcinoma
A Phase 2 study of sunitinib was conducted in patients who were refractory to prior cytokine therapy with interleukin-2 or IFN-α. Sixty-three patients received a starting dose of 50 mg sunitinib orally, once daily for 4 consecutive weeks followed by a 2-week rest period, to comprise a complete cycle of 6 weeks (Schedule 4/2). The primary efficacy endpoint was ORR, based on Response Evaluation Criteria in Solid Tumours (RECIST).
In this study the objective response rate was 36.5% (95% CI: 24.7%, 49.6%) and the median TTP was 37.7 weeks (95% CI: 24.0, 46.4).
A confirmatory, open-label, single-arm, multi-centre study evaluating the efficacy and safety of sunitinib was conducted in patients with MRCC who were refractory to prior cytokine therapy. One hundred and 6 patients received at least one 50 mg dose of sunitinib on Schedule 4/2.
The primary efficacy endpoint of this study was ORR. Secondary endpoints included TTP, duration of response (DR) and OS.
In this study the ORR was 35.8% (95% CI: 26.8%, 47.5 %). The median DR and OS had not yet been reached.
Pancreatic neuroendocrine tumours
A supportive Phase 2, open-label, multi-centre study evaluated the efficacy and safety of single-agent sunitinib 50 mg daily on Schedule 4/2 in patients with unresectable pNET. In a pancreatic islet cell tumour cohort of 66 patients, the primary endpoint of response rate was 17%.
A pivotal Phase 3, multi-centre, international, randomised, double-blind, placebo-controlled study of single-agent sunitinib was conducted in patients with unresectable pNET.
Patients were required to have documented progression, based on RECIST, within the prior 12 months and were randomised (1:1) to receive either 37.5 mg sunitinib once daily without a scheduled rest period (N = 86) or placebo (N = 85).
The primary objective was to compare PFS in patients receiving sunitinib versus patients receiving placebo. Other endpoints included OS, ORR, PROs, and safety.
Demographics were comparable between the sunitinib and placebo groups. Additionally, 49% of sunitinib patients had nonfunctioning tumours versus 52% of placebo patients and 92% of patients in both arms had liver metastases.
Use of somatostatin analogues was allowed in the study.
A total of 66% of sunitinib patients received prior systemic therapy compared with 72% of placebo patients. In addition, 24% of sunitinib patients had received somatostatin analogues compared with 22% of placebo patients.
A clinically significant advantage in investigator-assessed PFS for sunitinib over placebo was observed. The median PFS was 11.4 months for the sunitinib arm compared to 5.5 months for the placebo arm [hazard ratio: 0.418 (95% CI: 0.263, 0.662), p-value=0.0001]; similar results were observed when derived tumour response assessments based upon application of RECIST to investigator tumour measurements were used to determine disease progression, as shown in Table 4. A hazard ratio favouring sunitinib was observed in all subgroups of baseline characteristics evaluated, including an analysis by number of prior systemic therapies. A total of 29 patients in the sunitinib arm and 24 in the placebo arm had received no prior systemic treatment; among these patients, the hazard ratio for PFS was 0.365 (95% CI: 0.156, 0.857), p=0.0156. Similarly, among 57 patients in the sunitinib arm (including 28 with 1 prior systemic therapy and 29 with 2 or more prior systemic therapies) and 61 patients in the placebo arm (including 25 with 1 prior systemic therapy and 36 with 2 or more prior systemic therapies), the hazard ratio for PFS was 0.456 (95% CI: 0.264, 0.787), p = 0.0036.
A sensitivity analysis of PFS was conducted where progression was based upon investigator-reported tumour measurements and where all subjects censored for reasons other than study termination were treated as PFS events. This analysis provided a conservative estimate of the treatment effect of sunitinib and supported the primary analysis, demonstrating a hazard ratio of 0.507 (95% CI: 0.350, 0.733), p = 0.000193. The pivotal study in pancreatic NET was terminated prematurely at the recommendation of an independent drug monitoring committee and the primary endpoint was based upon investigator assessment, both of which may have affected the estimates of the treatment effect.
In order to rule out bias in the investigator-based assessment of PFS, a BICR of scans was performed; this review supported the investigator assessment, as shown in Table 4.
Table 4 - pNET efficacy results from the Phase 3 study
|
Efficacy parameter
|
Sutent
(N = 86)
|
Placebo
(N = 85)
|
Hazard Ratio
(95% CI)
|
p-value
|
|
Progression-free survival [median, months (95% CI)] by Investigator Assessment
|
11.4
(7.4, 19.8)
|
5.5
(3.6, 7.4)
|
0.418
(0.263, 0.662)
|
0.0001a
|
|
Progression-free survival [median, months (95% CI)] by derived tumour response assessment based upon application of RECIST to investigator tumour assessments
|
12.6
(7.4, 16.9)
|
5.4
(3.5, 6.0)
|
0.401
(0.252, 0.640)
|
0.000066a
|
|
Progression-free survival [median, months (95% CI)] by blinded independent central review of tumour assessments
|
12.6
(11.1, 20.6)
|
5.8
(3.8, 7.2)
|
0.315
(0.181, 0.546)
|
0.000015a
|
|
Overall survival [5 years follow-up] [median, months (95% CI)]
|
38.6
(25.6, 56.4)
|
29.1
(16.4, 36.8)
|
0.730
(0.504, 1.057)
|
0.0940a
|
|
Objective response rate [%, (95% CI)]
|
9.3
(3.2, 15.4)
|
0
|
NA
|
0.0066b
|
Abbreviations: CI=confidence interval; N=number of patients; NA=not applicable; pNET=pancreatic neuroendocrine tumours; RECIST=response evaluation criteria in solid tumours.
a 2-sided unstratified log-rank test
b Fisher's Exact test
Figure 1. Kaplan-Meier curve of PFS in the pNET Phase 3 study
Abbreviations: CI=confidence interval; N=number of patients; PFS=progression-free survival; pNET=pancreatic neuroendocrine tumours.
OS data were not mature at the time of the study closure [20.6 months (95% CI: 20.6, NR) for the sunitinib arm compared to NR (95% CI: 15.5, NR) for the placebo arm, hazard ratio: 0.409 (95% CI: 0.187, 0.894), p-value=0.0204]. There were 9 deaths in the sunitinib arm and 21 deaths in the placebo arm.
Upon disease progression, patients were unblinded and placebo patients were offered access to open-label sunitinib in a separate extension study. As a result of the early study closure, remaining patients were unblinded and offered access to open-label sunitinib in an extension study. A total of 59 out of 85 patients (69.4%) from the placebo arm crossed over to open-label sunitinib following disease progression or unblinding at study closure. OS observed after 5 years of follow-up in the extension study showed a hazard ratio of 0.730 (95% CI: 0.504, 1.057).
Results from the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-C30) showed that the overall global health-related quality of life and the 5 functioning domains (physical, role, cognitive, emotional, and social) were maintained for patients on sunitinib treatment as compared to placebo with limited adverse symptomatic effects.
A Phase 4 multinational, multi-centre, single-arm, open-label study evaluating the efficacy and safety of sunitinib was conducted in patients with progressive, advanced/metastatic, well-differentiated, unresectable pNET.
One hundred six patients (61 patients in the treatment-naïve cohort and 45 patients in the later-line cohort) received treatment with sunitinib orally at 37.5 mg once a day on a continuous daily dosing (CDD) schedule.
The investigator-assessed median PFS was 13.2 months, both in the overall population (95% CI: 10.9, 16.7) and in the treatment-naïve cohort (95% CI: 7.4, 16.8).
Paediatric population
Experience on the use of sunitinib in paediatric patients is limited.
A Phase 1 dose-escalation study of oral sunitinib was conducted in paediatric and young adult patients (age: 2-21 years) with refractory solid tumours, the majority of whom were enrolled with a primary diagnosis of brain tumour. Dose-limiting cardiotoxicity was observed in the first part of the study which was therefore amended to exclude patients with previous exposure to potentially cardiotoxic therapies (including anthracyclines) or cardiac radiation. In the second part of the study, including patients with prior anticancer therapy but without risk factors for cardiac toxicity, sunitinib was generally tolerable and clinically manageable at the dose of 15 mg/m2/day on Schedule 4/2. None of the subjects achieved complete response or partial response. Stable disease was observed in 6 patients (17%). One GIST patient was enrolled at the 15 mg/m2 dose level with no evidence of benefit. The observed adverse drug reactions were overall similar to those seen in adults.
A population PK and PK/PD analysis was conducted with the scope to extrapolate the PK and key safety and efficacy endpoints of sunitinib in paediatric patients with GIST (age: 6-17 years). This analysis was based on data collected from adults with GIST or solid tumours and from paediatric patients with solid tumours. Based on the modelling analyses, the younger age and lower body size did not appear to affect negatively the safety and efficacy responses to sunitinib plasma exposures. Sunitinib benefit/risk did not appear to be negatively affected by younger age or lower body size, and was mainly driven by its plasma exposure.
Based on the PK, safety, and efficacy trial simulation results, a starting dose of approximately 25 mg/m2/day on Schedule 4/2 in paediatric patients with GIST (ages: 6-11 and 12-17 years) is predicted to provide comparable sunitinib plasma exposures, and subsequently safety and efficacy to those in adult patients with GIST treated at 50 mg/day on Schedule 4/2.
The European Medicines Agency (EMA) has deferred the obligation to submit the results of the studies with Sutent in 1 or more subsets of the paediatric population in GIST.
The EMA has waived the obligation to submit the results of studies with Sutent in all subsets of the paediatric population for treatment of kidney and renal pelvis carcinoma (excluding nephroblastoma, nephroblastomatosis, clear cell sarcoma, mesoblastic nephroma, renal medullary carcinoma, and rhabdoid tumour of the kidney).
The EMA has waived the obligation to submit the results of the studies with Sutent in all subsets of the paediatric population for treatment of gastroenteropancreatic neuroendocrine tumours (excluding neuroblastoma, neuroganglioblastoma, and phaeochromocytoma).
Pharmacokinetic properties
The pharmacokinetics of sunitinib were evaluated in 135 healthy volunteers and 266 patients with solid tumours. The pharmacokinetics were similar in all solid tumours populations tested and in healthy volunteers.
In the dosing ranges of 25 to 100 mg, the area under the plasma concentration-time curve (AUC) and Cmax increase proportionally with dose. With repeated daily administration, sunitinib accumulates 3- to 4-fold and its primary active metabolite accumulates 7- to 10-fold. Steady-state concentrations of sunitinib and its primary active metabolite are achieved within 10 to 14 days. By Day 14, combined plasma concentrations of sunitinib and its active metabolite are 62.9-101 ng/ml, which are target concentrations predicted from preclinical data to inhibit receptor phosphorylation in vitro and result in tumour stasis/growth reduction in vivo. The primary active metabolite comprises 23% to 37% of the total exposure. No significant changes in the pharmacokinetics of sunitinib or the primary active metabolite are observed with repeated daily administration or with repeated cycles in the dosing schedules tested.
Absorption
After oral administration of sunitinib, Cmax are generally observed from 6 to 12 hours time to maximum concentration (tmax) postadministration.
Food has no effect on the bioavailability of sunitinib.
Distribution
In vitro, binding of sunitinib and its primary active metabolite to human plasma protein was 95% and 90%, respectively, with no apparent concentration dependence. The apparent volume of distribution (Vd) for sunitinib was large, 2230 L, indicating distribution into the tissues.
Metabolic interactions
The calculated in vitro Ki values for all cytochrome P450 (CYP) isoforms tested (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, and CYP4A9/11) indicated that sunitinib and its primary active metabolite are unlikely to induce metabolism, to any clinically relevant extent, of other active substances that may be metabolised by these enzymes.
Biotransformation
Sunitinib is metabolised primarily by CYP3A4, the CYP isoform which produces its primary active metabolite, desethyl sunitinib, which is then further metabolised by the same isoenzyme.
Co-administration of sunitinib with potent CYP3A4 inducers or inhibitors should be avoided because the plasma levels of sunitinib may be altered.
Elimination
Excretion is primarily via faeces (61%), with renal elimination of unchanged active substance and metabolites accounting for 16% of the administered dose. Sunitinib and its primary active metabolite were the major compounds identified in plasma, urine, and faeces, representing 91.5%, 86.4%, and 73.8% of radioactivity in pooled samples, respectively. Minor metabolites were identified in urine and faeces, but generally were not found in plasma. Total oral clearance (CL/F) was 34-62 L/h. Following oral administration in healthy volunteers, the elimination half-lives of sunitinib and its primary active desethyl metabolite are approximately 40-60 hours and 80-110 hours, respectively.
Co-administration with medicinal products that are BCRP inhibitors
In vitro, sunitinib is a substrate of the efflux transporter BCRP. In study A6181038 the co-administration of gefitinib, a BCRP inhibitor, did not result in a clinically relevant effect on the Cmax and AUC for sunitinib or total drug (sunitinib + metabolite). This study was a multi-centre, open-label, Phase 1/2 study examining the safety/tolerability, the maximum tolerated dose, and the antitumour activity of sunitinib in combination with gefitinib in subjects with MRCC. The PK of gefitinib (250 mg daily) and sunitinib (37.5 mg [Cohort 1, n=4] or 50 mg [Cohort 2, n=7] daily on a 4-weeks on followed by 2 weeks-off schedule) when co-administered was evaluated as a secondary study objective. Changes in sunitinib pharmacokinetic parameters were of no clinical significance and did not indicate any drug-drug interactions; however, considering the relatively low number of subjects (i.e. N=7+4) and the moderate-large interpatient variability in the pharmacokinetic parameters, caution needs to be taken when interpreting the pharmacokinetic drug-drug interaction findings from this study.
Special populations
Hepatic impairment
Sunitinib and its primary metabolite are mainly metabolised by the liver. Systemic exposures after a single dose of sunitinib were similar in subjects with mild or moderate (Child-Pugh Class A and B) hepatic impairment compared to subjects with normal hepatic function. Sutent was not studied in subjects with severe (Child-Pugh Class C) hepatic impairment.
Studies in cancer patients have excluded patients with ALT or AST > 2.5 x ULN (upper limit of normal) or > 5.0 x ULN if due to liver metastasis.
Renal impairment
Population pharmacokinetic analyses indicated that sunitinib apparent clearance (CL/F) was not affected by creatinine clearance (CrCl) within the range evaluated (42-347 ml/min).
Systemic exposures after a single dose of sunitinib were similar in subjects with severe renal impairment (CrClr < 30 ml/min) compared to subjects with normal renal function (CrCl > 80 ml/min). Although sunitinib and its primary metabolite were not eliminated through haemodialysis in subjects with ESRD, the total systemic exposures were lower by 47% for sunitinib and 31% for its primary metabolite compared to subjects with normal renal function.
Weight, performance status
Population pharmacokinetic analyses of demographic data indicate that no starting dose adjustments are necessary for weight or Eastern Cooperative Oncology Group (ECOG) performance status.
Gender
Available data indicate that females could have about 30% lower apparent clearance (CL/F) of sunitinib than males: this difference, however, does not necessitate starting dose adjustments.
Paediatric population
Experience on the use of sunitinib in paediatric patients is limited. Population PK analyses of a pooled dataset from adult patients with GIST and solid tumours and paediatric patients with solid tumours were completed. Stepwise covariate modelling analyses were performed to evaluate the effect of age and body size (total body weight or body surface area) as well as other covariates on important PK parameters for sunitinib and its active metabolite. Among age and body size related covariates tested, age was a significant covariate on apparent clearance of sunitinib (the younger the age of the paediatric patient, the lower the apparent clearance). Similarly, body surface area was a significant covariate on the apparent clearance of the active metabolite (the lower the body surface area, the lower the apparent clearance). Based on the final PK model trial simulation results, taking into account all the covariates effects, a sunitinib dose of 25 mg/m2/day in paediatric patients (ages: 6-11 and 12-17 years) with GIST is predicted to achieve comparable sunitinib plasma exposures to those in adult patients with GIST treated at 50 mg/day, on Schedule 4/2.
Date of revision of the text
02/2018
Marketing authorisation holder
Pfizer Ltd
Ramsgate Road
Sandwich, Kent CT13 9NJ
United Kingdom
Special precautions for storage
This medicinal product does not require any special storage conditions.
Nature and contents of container
High-density polyethylene (HDPE) bottle with a polypropylene closure containing 30 hard capsules.
Poly(chlorotrifluoroethylene)/PVC transparent perforated unit dose blister with aluminium foil coated with heat seal lacquer containing 28 x 1 hard capsules.
Not all pack sizes may be marketed.
Marketing authorisation number(s)
Sutent 12.5 mg hard capsules
EU/1/06/347/001
EU/1/06/347/004
Sutent 25 mg hard capsules
EU/1/06/347/002
EU/1/06/347/005
Sutent 37.5 mg hard capsules
EU/1/06/347/007
EU/1/06/347/008
Sutent 50 mg hard capsules
EU/1/06/347/003
EU/1/06/347/006
Fertility, pregnancy and lactation
Contraception in males and females
Women of childbearing potential should be advised to use effective contraception and avoid becoming pregnant while receiving treatment with Sutent.
Pregnancy
There are no studies in pregnant women using sunitinib. Studies in animals have shown reproductive toxicity including foetal malformations. Sutent should not be used during pregnancy or in women not using effective contraception, unless the potential benefit justifies the potential risk to the foetus. If Sutent is used during pregnancy or if the patient becomes pregnant while on treatment with Sutent, the patient should be apprised of the potential hazard to the foetus.
Breast-feeding
Sunitinib and/or its metabolites are excreted in rat milk. It is not known whether sunitinib or its primary active metabolite is excreted in human milk. Because active substances are commonly excreted in human milk and because of the potential for serious adverse reactions in breast-feeding infants, women should not breast-feed while taking Sutent.
Fertility
Based on nonclinical findings, male and female fertility may be compromised by treatment with sunitinib.
Special warnings and precautions for use
Co-administration with potent CYP3A4 inducers should be avoided because it may decrease sunitinib plasma concentration.
Co-administration with potent CYP3A4 inhibitors should be avoided because it may increase the plasma concentration of sunitinib.
Skin and tissue disorders
Patients should be advised that depigmentation of the hair or skin may occur during treatment with sunitinib. Other possible dermatological effects may include dryness, thickness or cracking of the skin, blisters, or rash on the palms of the hands and soles of the feet.
The above reactions were not cumulative, were typically reversible, and generally did not result in treatment discontinuation. Cases of pyoderma gangrenosum, generally reversible after discontinuation of sunitinib, have been reported. Severe cutaneous reactions have been reported, including cases of erythema multiforme (EM), cases suggestive of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), some of which were fatal. If signs or symptoms of SJS, TEN, or EM (e.g., progressive skin rash often with blisters or mucosal lesions) are present, sunitinib treatment should be discontinued. If the diagnosis of SJS or TEN is confirmed, treatment must not be restarted. In some cases of suspected EM, patients tolerated the reintroduction of sunitinib therapy at a lower dose after resolution of the reaction; some of these patients also received concomitant treatment with corticosteroids or antihistamines.
Haemorrhage and tumour bleeding
Haemorrhagic events, some of which were fatal, reported in clinical studies with sunitinib and during postmarketing surveillance have included gastrointestinal, respiratory, urinary tract, and brain haemorrhages.
Routine assessment of bleeding events should include complete blood counts and physical examination.
Epistaxis was the most common haemorrhagic adverse reaction, having been reported for approximately half of the patients with solid tumours who experienced haemorrhagic events. Some of the epistaxis events were severe, but very rarely fatal.
Events of tumour haemorrhage, sometimes associated with tumour necrosis, have been reported; some of these haemorrhagic events were fatal.
Tumour haemorrhage may occur suddenly, and in the case of pulmonary tumours, may present as severe and life-threatening haemoptysis or pulmonary haemorrhage. Cases of pulmonary haemorrhage, some with a fatal outcome, have been observed in clinical trials and have been reported in postmarketing experience in patients treated with sunitinib for MRCC, GIST, and lung cancer. Sutent is not approved for use in patients with lung cancer.
Patients receiving concomitant treatment with anticoagulants (e.g., warfarin, acenocoumarole) may be periodically monitored by complete blood counts (platelets), coagulation factors (PT/INR), and physical examination.
Gastrointestinal disorders
Diarrhoea, nausea/vomiting, abdominal pain, dyspepsia, and stomatitis/oral pain were the most commonly reported gastrointestinal adverse reactions; oesophagitis events have been also reported.
Supportive care for gastrointestinal adverse reactions requiring treatment may include medicinal products with antiemetic, antidiarrhoeal, or antacid properties.
Serious, sometimes fatal gastrointestinal complications including gastrointestinal perforation were reported in patients with intra-abdominal malignancies treated with sunitinib.
Hypertension
Hypertension has been reported in association with sunitinib, including severe hypertension (> 200 mmHg systolic or 110 mmHg diastolic). Patients should be screened for hypertension and controlled as appropriate. Temporary suspension is recommended in patients with severe hypertension that is not controlled with medical management. Treatment may be resumed once hypertension is appropriately controlled .
Haematological disorders
Decreased absolute neutrophil counts and decreased platelet counts were reported in association with sunitinib. The above events were not cumulative, were typically reversible, and generally did not result in treatment discontinuation. None of these events in the Phase 3 studies were fatal, but rare fatal haematological events, including haemorrhage associated with thrombocytopenia and neutropenic infections, have been reported during postmarketing surveillance.
Anaemia has been observed to occur early as well as late during treatment with sunitinib.
Complete blood counts should be performed at the beginning of each treatment cycle for patients receiving treatment with sunitinib.
Cardiac disorders
Cardiovascular events, including heart failure, cardiomyopathy, left ventricular ejection fraction decline to below the lower limit of normal, myocarditis, myocardial ischaemia and myocardial infarction, some of which were fatal, have been reported in patients treated with sunitinib. These data suggest that sunitinib increases the risk of cardiomyopathy. No specific additional risk factors for sunitinib-induced cardiomyopathy apart from the drug-specific effect have been identified in the treated patients. Use sunitinib with caution in patients who are at risk for, or who have a history of, these events.
Patients who presented with cardiac events within 12 months prior to sunitinib administration, such as myocardial infarction (including severe/unstable angina), coronary/peripheral artery bypass graft, symptomatic congestive heart failure (CHF), cerebrovascular accident or transient ischaemic attack, or pulmonary embolism were excluded from all sunitinib clinical studies. It is unknown whether patients with these concomitant conditions may be at a higher risk of developing sunitinib-related left ventricular dysfunction.
Physicians are advised to weigh this risk against the potential benefits of sunitinib. Patients should be carefully monitored for clinical signs and symptoms of CHF while receiving sunitinib especially patients with cardiac risk factors and/or history of coronary artery disease. Baseline and periodic evaluations of LVEF should also be considered while the patient is receiving sunitinib. In patients without cardiac risk factors, a baseline evaluation of ejection fraction should be considered.
In the presence of clinical manifestations of CHF, discontinuation of sunitinib is recommended. The administration of sunitinib should be interrupted and/or the dose reduced in patients without clinical evidence of CHF but with an ejection fraction < 50% and > 20% below baseline.
QT interval prolongation
Prolongation of QT interval and Torsade de pointes have been observed in sunitinib-exposed patients. QT interval prolongation may lead to an increased risk of ventricular arrhythmias including Torsade de pointes.
Sunitinib should be used with caution in patients with a known history of QT interval prolongation, patients who are taking antiarrhythmics or medicinal products that can prolong QT interval, or patients with relevant pre-existing cardiac disease, bradycardia, or electrolyte disturbances. Concomitant administration of sunitinib with potent CYP3A4 inhibitors should be limited because of the possible increase in sunitinib plasma concentrations.
Venous thromboembolic events
Treatment-related venous thromboembolic events were reported in patients who received sunitinib including deep venous thrombosis and pulmonary embolism. Cases of pulmonary embolism with fatal outcome have been observed in postmarketing surveillance.
Arterial thromboembolic events
Cases of arterial thromboembolic events (ATE), sometimes fatal, have been reported in patients treated with sunitinib. The most frequent events included cerebrovascular accident, transient ischaemic attack, and cerebral infarction. Risk factors associated with ATE, in addition to the underlying malignant disease and age > 65 years, included hypertension, diabetes mellitus, and prior thromboembolic disease.
Thrombotic microangiopathy (TMA)
The diagnosis of TMA, including thrombotic thrombocytopaenic purpura (TTP) and haemolytic uraemic syndrome (HUS), sometimes leading to renal failure or a fatal outcome, should be considered in the occurrence of haemolytic anaemia, thrombocytopenia, fatigue, fluctuating neurological manifestation, renal impairment, and fever. Sunitinib therapy should be discontinued in patients who develop TMA and prompt treatment is required. Reversal of the effects of TMA has been observed after treatment discontinuation.
Thyroid dysfunction
Baseline laboratory measurement of thyroid function is recommended in all patients. Patients with pre-existing hypothyroidism or hyperthyroidism should be treated as per standard medical practice prior to the start of sunitinib treatment. During sunitinib treatment, routine monitoring of thyroid function should be performed every 3 months. In addition, patients should be observed closely for signs and symptoms of thyroid dysfunction during treatment, and patients who develop any signs and/or symptoms suggestive of thyroid dysfunction should have laboratory testing of thyroid function performed as clinically indicated. Patients who develop thyroid dysfunction should be treated as per standard medical practice.
Hypothyroidism has been observed to occur early as well as late during treatment with sunitinib.
Pancreatitis
Increases in serum lipase and amylase activities were observed in patients with various solid tumours who received sunitinib. Increases in lipase activities were transient and were generally not accompanied by signs or symptoms of pancreatitis in subjects with various solid tumours.
Cases of serious pancreatic events, some with fatal outcome, have been reported. If symptoms of pancreatitis are present, patients should have sunitinib discontinued and be provided with appropriate supportive care.
Hepatotoxicity
Hepatotoxicity has been observed in patients treated with sunitinib. Cases of hepatic failure, some with a fatal outcome, were observed in < 1% of solid tumour patients treated with sunitinib. Monitor liver function tests (alanine transaminase [ALT], aspartate transaminase [AST], bilirubin levels) before initiation of treatment, during each cycle of treatment, and as clinically indicated. If signs or symptoms of hepatic failure are present, sunitinib should be discontinued and appropriate supportive care should be provided.
Renal function
Cases of renal impairment, renal failure and/or acute renal failure, in some cases with fatal outcome, have been reported.
Risk factors associated with renal impairment/failure in patients receiving sunitinib included, in addition to underlying RCC, older age, diabetes mellitus, underlying renal impairment, cardiac failure, hypertension, sepsis, dehydration/hypovolaemia, and rhabdomyolysis.
The safety of continued sunitinib treatment in patients with moderate to severe proteinuria has not been systematically evaluated.
Cases of proteinuria and rare cases of nephrotic syndrome have been reported. Baseline urinalysis is recommended, and patients should be monitored for the development or worsening of proteinuria. Discontinue sunitinib in patients with nephrotic syndrome.
Fistula
If fistula formation occurs, sunitinib treatment should be interrupted. Limited information is available on the continued use of sunitinib in patients with fistulae.
Impaired wound healing
Cases of impaired wound healing have been reported during sunitinib therapy.
No formal clinical studies of the effect of sunitinib on wound healing have been conducted. Temporary interruption of sunitinib therapy is recommended for precautionary reasons in patients undergoing major surgical procedures. There is limited clinical experience regarding the timing of reinitiation of therapy following major surgical intervention. Therefore, the decision to resume sunitinib therapy following a major surgical intervention should be based upon clinical judgment of recovery from surgery.
Osteonecrosis of the jaw (ONJ)
Cases of ONJ have been reported in patients treated with Sutent. The majority of cases were reported in patients who had received prior or concomitant treatment with intravenous bisphosphonates, for which ONJ is an identified risk. Caution should therefore be exercised when Sutent and intravenous bisphosphonates are used either simultaneously or sequentially.
Invasive dental procedures are also an identified risk factor. Prior to treatment with Sutent, a dental examination and appropriate preventive dentistry should be considered. In patients who have previously received or are receiving intravenous bisphosphonates, invasive dental procedures should be avoided if possible.
Hypersensitivity/angioedema
If angioedema due to hypersensitivity occurs, sunitinib treatment should be interrupted and standard medical care provided.
Seizures
In clinical studies of sunitinib and from postmarketing surveillance, seizures have been reported. Patients with seizures and signs/symptoms consistent with posterior reversible leukoencephalopathy syndrome (RPLS), such as hypertension, headache, decreased alertness, altered mental functioning and visual loss, including cortical blindness, should be controlled with medical management including control of hypertension. Temporary suspension of sunitinib is recommended; following resolution, treatment may be resumed at the discretion of the treating physician.
Tumour lysis syndrome (TLS)
Cases of TLS, some fatal, have been rarely observed in clinical trials and have been reported in postmarketing surveillance in patients treated with sunitinib. Risk factors for TLS include high tumour burden, pre-existing chronic renal insufficiency, oliguria, dehydration, hypotension, and acidic urine. These patients should be monitored closely and treated as clinically indicated, and prophylactic hydration should be considered.
Infections
Serious infections, with or without neutropenia, including some with a fatal outcome, have been reported. Uncommon cases of necrotising fasciitis, including of the perineum, sometimes fatal, have been reported.
Sunitinib therapy should be discontinued in patients who develop necrotising fasciitis, and appropriate treatment should be promptly initiated.
Hypoglycaemia
Decreases in blood glucose, in some cases clinically symptomatic and requiring hospitalisation due to loss of consciousness, have been reported during sunitinib treatment. In case of symptomatic hypoglycaemia, sunitinib should be temporarily interrupted. Blood glucose levels in diabetic patients should be checked regularly in order to assess if antidiabetic medicinal product's dosage needs to be adjusted to minimise the risk of hypoglycaemia.
Effects on ability to drive and use machines
Sutent has minor influence on the ability to drive and use machines. Patients should be advised that they may experience dizziness during treatment with sunitinib.
Dosage (Posology) and method of administration
Therapy with Sutent should be initiated by a physician experienced in the administration of anticancer agents.
Posology
For GIST and MRCC, the recommended dose of Sutent is 50 mg taken orally once daily, for 4 consecutive weeks, followed by a 2-week rest period (Schedule 4/2) to comprise a complete cycle of 6 weeks.
For pNET, the recommended dose of Sutent is 37.5 mg taken orally once daily without a scheduled rest period.
Dose adjustments
Safety and tolerability
For GIST and MRCC, dose modifications in 12.5 mg steps may be applied based on individual safety and tolerability. Daily dose should not exceed 75 mg nor be decreased below 25 mg.
For pNET, dose modification in 12.5 mg steps may be applied based on individual safety and tolerability. The maximum dose administered in the Phase 3 pNET study was 50 mg daily.
Dose interruptions may be required based on individual safety and tolerability.
CYP3A4 inhibitors/inducers
Co-administration of sunitinib with potent CYP3A4 inducers, such as rifampicin, should be avoided. If this is not possible, the dose of sunitinib may need to be increased in 12.5 mg steps (up to 87.5 mg per day for GIST and MRCC or 62.5 mg per day for pNET) based on careful monitoring of tolerability.
Co-administration of sunitinib with potent CYP3A4 inhibitors, such as ketoconazole, should be avoided. If this is not possible, the dose of sunitinib may need to be reduced to a minimum of 37.5 mg daily for GIST and MRCC or 25 mg daily for pNET, based on careful monitoring of tolerability.
Selection of an alternative concomitant medicinal product with no or minimal potential to induce or inhibit CYP3A4 should be considered.
Special populations
Paediatric population
The safety and efficacy of Sutent in patients below 18 years of age have not been established.
1, and 5.2 but no recommendation on a posology can be made.
Elderly
Approximately one-third of the patients in clinical studies who received sunitinib were 65 years of age or over. No significant differences in safety or efficacy were observed between younger and older patients.
Hepatic impairment
No starting dose adjustment is recommended when administering sunitinib to patients with mild or moderate (Child-Pugh class A and B) hepatic impairment. Sunitinib has not been studied in subjects with severe (Child-Pugh class C) hepatic impairment and therefore its use in patients with severe hepatic impairment cannot be recommended.
Renal impairment
No starting dose adjustment is required when administering sunitinib to patients with renal impairment (mild-severe) or with end-stage renal disease (ESRD) on haemodialysis. Subsequent dose adjustments should be based on individual safety and tolerability.
Method of administration
Sutent is for oral administration. It may be taken with or without food.
If a dose is missed, the patient should not be given an additional dose. The patient should take the usual prescribed dose on the following day.
Special precautions for disposal and other handling
No special requirements.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Date of first authorisation/renewal of the authorisation
Date of first authorisation: 19 July 2006
Date of latest renewal: 9 November 2016
Interaction with other medicinal products and other forms of interaction
Interaction studies have only been performed in adults.
Medicinal products that may increase sunitinib plasma concentrations
Effect of CYP3A4 inhibitors
In healthy volunteers, concomitant administration of a single dose of sunitinib with the potent CYP3A4 inhibitor ketoconazole resulted in an increase of the combined [sunitinib + primary metabolite] maximum concentration (Cmax) and area under the curve (AUC0-∞) values of 49% and 51%, respectively.
Administration of sunitinib with potent CYP3A4 inhibitors (e.g., ritonavir, itraconazole, erythromycin, clarithromycin, grapefruit juice) may increase sunitinib concentrations.
Combination with CYP3A4 inhibitors should therefore be avoided, or the selection of an alternate concomitant medicinal product with no or minimal potential to inhibit CYP3A4 should be considered.
If this is not possible, the dose of Sutent may need to be reduced to a minimum of 37.5 mg daily for GIST and MRCC or 25 mg daily for pNET, based on careful monitoring of tolerability.
Effect of Breast Cancer Resistance Protein (BCRP) inhibitors
Limited clinical data are available on the interaction between sunitinib and BCRP inhibitors and the possibility of an interaction between sunitinib and other BCRP inhibitors cannot be excluded.
Medicinal products that may decrease sunitinib plasma concentrations
Effect of CYP3A4 inducers
In healthy volunteers, concomitant administration of a single dose of sunitinib with the CYP3A4 inducer rifampicin resulted in a reduction of the combined [sunitinib + primary metabolite] Cmax and AUC0-∞ values of 23% and 46%, respectively.
Administration of sunitinib with potent CYP3A4 inducers (e.g., dexamethasone, phenytoin, carbamazepine, rifampicin, phenobarbital or herbal preparations containing St. John's Wort/Hypericum perforatum) may decrease sunitinib concentrations.
Combination with CYP3A4 inducers should therefore be avoided, or selection of an alternate concomitant medicinal product, with no or minimal potential to induce CYP3A4 should be considered.
If this is not possible, the dose of Sutent may need to be increased in 12.5 mg increments (up to 87.5 mg per day for GIST and MRCC or 62.5 mg per day for pNET), based on careful monitoring of tolerability.