Starlix
Drugs with the same active ingredients
Overdose
In a clinical study in patients, Starlix was administered in increasing doses up to 720 mg a day for 7 days and was well tolerated. There is no experience of an overdose of Starlix in clinical trials. However, an overdose may result in an exaggerated glucose-lowering effect, with the development of hypoglycaemic symptoms. Hypoglycaemic symptoms without loss of consciousness or neurological findings should be treated with oral glucose and adjustments in dosage and/or meal patterns. Severe hypoglycaemic reactions with coma, seizure or other neurological symptoms should be treated with intravenous glucose. As nateglinide is highly protein-bound, dialysis is not an efficient means of removing it from the blood.
Shelf life
3 years
Contraindications
- Type 1 diabetes (C-peptide negative)
- Diabetic ketoacidosis, with or without coma
- Pregnancy and breast-feeding
- Severe hepatic impairment
Incompatibilities
Not applicable
List of excipients
Lactose monohydrate
Cellulose, microcrystalline
Povidone
Croscarmellose sodium
Magnesium stearate
Yellow iron oxide (E172)
Hypromellose
Titanium dioxide (E171)
Talc
Macrogol
Silica, colloidal anhydrous
Pharmaceutical form
Film-coated tablet
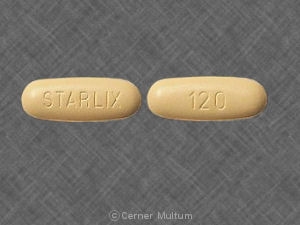
120 mg yellow, ovaloid tablets with “STARLIX†marked on one side and “120†on the other.
Undesirable effects
Based on the experience with nateglinide and with other hypoglycaemic agents, the following adverse reactions have been seen. Frequencies are defined as: very common (>1/10); common (>1/100 to <1/10); uncommon (>1/1,000 to <1/100); rare (>1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data).
Hypoglycaemia
As with other antidiabetic agents, symptoms suggestive of hypoglycaemia have been observed after administration of nateglinide. These symptoms included sweating, trembling, dizziness, increased appetite, palpitations, nausea, fatigue, and weakness. These were generally mild in nature and easily handled by intake of carbohydrates when necessary. In completed clinical trials, symptoms of hypoglycaemia were reported in 10.4% with nateglinide monotherapy, 14.5% with nateglinide+metformin combination, 6.9% with metformin alone, 19.8% with glibenclamide alone, and 4.1% with placebo.Immune system disorders
Rare: Hypersensitivity reactions such as rash, itching and urticaria.
Metabolism and nutrition disorders
Common: Symptoms suggestive of hypoglycaemia.
Gastrointestinal disorders
Common: Abdominal pain, diarrhoea, dyspepsia, nausea.
Uncommon: Vomiting.
Hepatobiliary disorders
Rare: Elevations in liver enzymes.
Other events
Other adverse events observed in clinical studies were of a similar incidence in Starlix-treated and placebo-treated patients.
Post-marketing data revealed very rare cases of erythema multiforme.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential and toxicity to fertility and post-natal development. Nateglinide was not teratogenic in rats. In rabbits, embryonic development was adversely affected and the incidence of gallbladder agenesis or small gallbladder was increased at doses of 300 and 500 mg/kg (approximately 24 and 28 times the human therapeutic exposure with a maximum recommended nateglinide dose of 180 mg, three times daily before meals), but not at 150 mg/kg (approximately 17 times the human therapeutic exposure with a maximum recommended nateglinide dose of 180 mg, three times daily before meals).
Therapeutic indications
Nateglinide is indicated for combination therapy with metformin in type 2 diabetic patients inadequately controlled despite a maximally tolerated dose of metformin alone.
Pharmacotherapeutic group
D-phenylalanine derivative, ATC code: A10 BX 03Pharmacodynamic properties
Pharmacotherapeutic group: D-phenylalanine derivative, ATC code: A10 BX 03
Nateglinide is an amino acid (phenylalanine) derivative, which is chemically and pharmacologically distinct from other antidiabetic agents. Nateglinide is a rapid, short-acting oral insulin secretagogue. Its effect is dependent on functioning beta cells in the pancreas islets.
Early insulin secretion is a mechanism for the maintenance of normal glycaemic control. Nateglinide, when taken before a meal, restores early or first phase insulin secretion, which is lost in patients with type 2 diabetes, resulting in a reduction in post-meal glucose and HbA1c.
Nateglinide closes ATP-dependent potassium channels in the beta-cell membrane with characteristics that distinguish it from other sulphonylurea receptor ligands. This depolarises the beta cell and leads to an opening of the calcium channels. The resulting calcium influx enhances insulin secretion. Electrophysiological studies demonstrate that nateglinide has 45-300-fold selectivity for pancreatic beta cell versus cardiovascular K+ATP channels.
In type 2 diabetic patients, the insulinotropic response to a meal occurs within the first 15 minutes following an oral dose of nateglinide. This results in a blood-glucose-lowering effect throughout the meal period. Insulin levels return to baseline within 3 to 4 hours, reducing post-meal hyperinsulinaemia.
Nateglinide-induced insulin secretion by pancreatic beta cells is glucose-sensitive, such that less insulin is secreted as glucose levels fall. Conversely, the coadministration of food or a glucose infusion results in an enhancement of insulin secretion.
In combination with metformin, which mainly affected fasting plasma glucose, the effect of nateglinide on HbA1c was additive compared to either agent alone.
Nateglinide efficacy was inferior to that of metformin in monotherapy (decrease in HbA1c (%) with metformin 500 mg three times daily monotherapy: -1.23 [95% CI: -1.48; -0.99] and with nateglinide 120 mg three times daily monotherapy -0.90 [95% CI: -1.14; -0.66]).
The efficacy of nateglinide in combination with metformin has been compared to the combination of gliclazide plus metformin in a 6-month randomised, double-blind trial in 262 patients using a superiority design. The decrease from baseline in HbA1c was -0.41% in the nateglinide plus metformin group and -0.57% in the gliclazide plus metformin group (difference 0.17%, [95% CI -0.03, 0.36]). Both treatments were well tolerated.
An outcome study has not been conducted with nateglinide, therefore the long-term benefits associated with improved glycaemic control have not been demonstrated.
Pharmacokinetic properties
Absorption
Nateglinide is rapidly absorbed following oral administration of Starlix tablets prior to a meal, with mean peak drug concentration generally occurring in less than 1 hour. Nateglinide is rapidly and almost completely (> 90%) absorbed from an oral solution. Absolute oral bioavailability is estimated to be 72%. In type 2 diabetic patients given Starlix over the dose range 60 to 240 mg before three meals per day for one week, nateglinide showed linear pharmacokinetics for both AUC and Cmax, and tmax was independent of dose.
Distribution
The steady-state volume of distribution of nateglinide based on intravenous data is estimated to be approximately 10 litres. In vitro studies show that nateglinide is extensively bound (97-99%) to serum proteins, mainly serum albumin and to a lesser extent alpha1-acid glycoprotein. The extent of serum protein binding is independent of drug concentration over the test range of 0.1-10 μg Starlix/ml.
Biotransformation
Nateglinide is extensively metabolised. The main metabolites found in humans result from hydroxylation of the isopropyl side-chain, either on the methine carbon, or one of the methyl groups; activity of the main metabolites is about 5-6 and 3 times less potent than nateglinide, respectively. Minor metabolites identified were a diol, an isopropene and acyl glucuronide(s) of nateglinide; only the isopropene minor metabolite possesses activity, which is almost as potent as nateglinide. Data available from both in vitro and in vivo experiments indicate that nateglinide is predominantly metabolised by CYP2C9 with involvement of CYP3A4 to a smaller extent.
Elimination
Nateglinide and its metabolites are rapidly and completely eliminated. Most of the [14C] nateglinide is excreted in the urine (83%), with an additional 10% eliminated in the faeces. Approximately 75% of the administered [14C] nateglinide is recovered in the urine within six hours post-dose. Approximately 6-16% of the administered dose was excreted in the urine as unchanged drug. Plasma concentrations decline rapidly and the elimination half-life of nateglinide typically averaged 1.5 hours in all studies of Starlix in volunteers and type 2 diabetic patients. Consistent with its short elimination half-life, there is no apparent accumulation of nateglinide upon multiple dosing with up to 240 mg three times daily.
Food effect
When given post-prandially, the extent of nateglinide absorption (AUC) remains unaffected. However, there is a delay in the rate of absorption characterised by a decrease in Cmax and a delay in time to peak plasma concentration (tmax). It is recommended that Starlix be administered prior to meals. It is usually taken immediately (1 minute) before a meal but may be taken up to 30 minutes before meals.
Special populations
Elderly
Age did not influence the pharmacokinetic properties of nateglinide.
Hepatic impairment
The systemic availability and half-life of nateglinide in non-diabetic subjects with mild to moderate hepatic impairment did not differ to a clinically significant degree from those in healthy subjects.
Renal impairment
The systemic availability and half-life of nateglinide in diabetic patients with mild, moderate (creatinine clearance 31-50 ml/min) and severe (creatinine clearance 15-30 ml/min) renal impairment (not undergoing dialysis) did not differ to a clinically significant degree from those in healthy subjects. There is a 49% decrease in Cmax of nateglinide in dialysis-dependent diabetic patients. The systemic availability and half-life in dialysis-dependent diabetic patients was comparable with healthy subjects. Although safety was not compromised in this population dose adjustment may be required in view of low Cmax.
Gender
No clinically significant differences in nateglinide pharmacokinetics were observed between men and women.
Date of revision of the text
16 June 2015
Marketing authorisation holder
Novartis Europharm Limited
Frimley Business Park
Camberley GU16 7SR
United Kingdom
Special precautions for storage
Do not store above 30°C.
Store in the original package.
Nature and contents of container
Blisters: PVC/PE/PVDC moulded foil with aluminium lidding foil.
Packs contain 12, 24, 30, 60, 84, 120 and 360 tablets.
Not all pack sizes may be marketed.
Marketing authorisation number(s)
EU/1/01/174/008-014
Fertility, pregnancy and lactation
Pregnancy
Studies in animals have shown developmental toxicity. There is no experience in pregnant women, therefore the safety of Starlix in pregnant women cannot be assessed. Starlix, like other oral antidiabetic agents, must not be used in pregnancy.
Breast-feeding
Nateglinide is excreted in the milk following a peroral dose to lactating rats. Although it is not known whether nateglinide is excreted in human milk, the potential for hypoglycaemia in breast-fed infants may exist and therefore nateglinide should not be used in lactating women.
Special warnings and precautions for use
General
Nateglinide should not be used in monotherapy.
Like other insulin secretagogues, nateglinide is capable of producing hypoglycaemia.
Hypoglycaemia has been observed in patients with type 2 diabetes on diet and exercise, and in those treated with oral antidiabetic agents. Elderly, malnourished patients and those with adrenal or pituitary insufficiency or severe renal impairment are more susceptible to the glucose-lowering effect of these treatments. The risk of hypoglycaemia in type 2 diabetic patients may be increased by strenuous physical exercise, or ingestion of alcohol.
Symptoms of hypoglycaemia (unconfirmed by blood glucose levels) were observed in patients whose baseline HbA1c was close to the therapeutic target (HbA1c <7.5%).
Combination with metformin is associated with an increased risk of hypoglycaemia compared to monotherapy.
Hypoglycaemia may be difficult to recognise in subjects receiving beta blockers.
When a patient stabilised on any oral hypoglycaemic agent is exposed to stress such as fever, trauma, infection or surgery, a loss of glycaemic control may occur. At such times, it may be necessary to discontinue oral hypoglycaemic treatment and replace it with insulin on a temporary basis.
Starlix contains lactose monohydrate. Patients with rare hereditary problems of galactose intolerance, of the Lapp lactase deficiency or of glucose-galactose malabsorption should not take this medicine.
Special populations
Nateglinide should be used with caution in patients with moderate hepatic impairment.
No clinical studies have been conducted in patients with severe hepatic impairment or children and adolescents. Treatment is therefore not recommended in these patient groups.
Effects on ability to drive and use machines
The effect of Starlix on the ability to drive or operate machinery has not been studied.
Patients should be advised to take precautions to avoid hypoglycaemia whilst driving. This is particularly important in those who have reduced or absent awareness of the warning signs of hypoglycaemia or have frequent episodes of hypoglycaemia. The advisability of driving should be considered in these circumstances.
Dosage (Posology) and method of administration
Posology
Nateglinide should be taken within 1 to 30 minutes before meals (usually breakfast, lunch and dinner).
The dosage of nateglinide should be determined by the physician according to the patient's requirements.
The recommended starting dose is 60 mg three times daily before meals, particularly in patients who are near goal HbA1c. This may be increased to 120 mg three times daily.
Dose adjustments should be based on periodic glycosylated haemoglobin (HbA1c) measurements. Since the primary therapeutic effect of Starlix is to reduce mealtime glucose, (a contributor to HbA1c), the therapeutic response to Starlix may also be monitored with 1-2 hour post-meal glucose.
The recommended maximum daily dose is 180 mg three times daily to be taken before the three main meals.
Special populations
Elderly
The clinical experience in patients over 75 years of age is limited.
Paediatric population
There are no data available on the use of nateglinide in patients under 18 years of age, and therefore its use in this age group is not recommended.
Patients with hepatic impairment
No dose adjustment is necessary for patients with mild to moderate hepatic impairment. As patients with severe liver disease were not studied, nateglinide is contraindicated in this group.
Patients with renal impairment
No dose adjustment is necessary in patients with mild to moderate renal impairment. Although there is a 49% decrease in Cmax of nateglinide in dialysis patients, the systemic availability and half-life in diabetic subjects with moderate to severe renal insufficiency (creatinine clearance 15-50 ml/min) was comparable between renal subjects requiring haemodialysis and healthy subjects. Although safety was not compromised in this population dose adjustment may be required in view of low Cmax.
Others
In debilitated or malnourished patients the initial and maintenance dosage should be conservative and careful titration is required to avoid hypoglycaemic reactions.
Special precautions for disposal and other handling
No special requirements
Date of first authorisation/renewal of the authorisation
Date of first authorisation: 03 April 2001
Date of latest renewal: 03 April 2006
Interaction with other medicinal products and other forms of interaction
A number of medicinal products influence glucose metabolism and possible interactions should therefore be taken into account by the physician:
The following agents may enhance the hypoglycaemic effect of nateglinide: angiotensin-converting enzyme inhibitors (ACEI), non-steroidal anti-inflammatory agents, salicylates, monoamine oxidase inhibitors, non-selective beta-adrenergic-blocking agents and anabolic hormones (e.g. methandrostenolone).
The following agents may reduce the hypoglycaemic effect of nateglinide: diuretics, corticosteroids, beta2 agonists, somatropin, somatostatin analogues (e.g. lanreotide, octreotide), rifampin, phenytoin and St John's wort.
When these medicinal products - that enhance or reduce the hypoglycaemic effect of nateglinide - are administered to or withdrawn from patients receiving nateglinide, the patient should be observed closely for changes in glycaemic control.
Data available from both in vitro and in vivo experiments indicate that nateglinide is predominantly metabolised by CYP2C9 with involvement of CYP3A4 to a smaller extent.
In an interaction trial with sulfinpyrazone, a CYP2C9 inhibitor, a modest increase in nateglinide AUC (~28%) was observed in healthy volunteers, with no changes in the mean Cmax and elimination half-life. A more prolonged effect and possibly a risk of hypoglycaemia cannot be excluded in patients when nateglinide is co-administered with CYP2C9 inhibitors.
Particular caution is recommended when nateglinide is co-administered with other more potent inhibitors of CYP2C9 (e.g. fluconazole, gemfibrozil or sulfinpyrazone), or in patients known to be poor metabolisers for CYP2C9.
Interaction studies with a 3A4 inhibitor have not been carried out in vivo.
In vivo, nateglinide has no clinically relevant effect on the pharmacokinetics of medicinal products metabolised by CYP2C9 and CYP3A4. The pharmacokinetics of warfarin (a substrate for CYP3A4 and CYP2C9), diclofenac (a substrate for CYP2C9), and digoxin were unaffected by coadministration with nateglinide. Conversely, these medicinal products had no effect on the pharmacokinetics of nateglinide. Thus, no dosage adjustment is required for digoxin, warfarin or other drugs that are CYP2C9 or CYP3A4 substrates upon coadministration with Starlix. Similarly, there was no clinically significant pharmacokinetic interaction of Starlix with other oral antidiabetic agents such as metformin or glibenclamide.
Nateglinide has shown a low potential for protein displacement in in vitro studies.