Soriatane
Drugs with the same active ingredients
Overdose
In the event of acute overdosage, SORIATANE must be withdrawn at once. Symptoms of overdose are identical to acute hypervitaminosis A (e.g., headache and vertigo). The acute oral toxicity (LD50) of acitretin in both mice and rats was greater than 4,000 mg per kg.
In one reported case of overdose, a 32-year-old male with Darier’s disease took 21 x 25-mg capsules (525-mg single dose). He vomited several hours later but experienced no other ill effects.
All female patients of childbearing potential who have taken an overdose of SORIATANE must:
- Have a pregnancy test at the time of overdose; 2) Be counseled as per the boxed
Contraindications
Pregnancy Category X(See boxed CONTRAINDICATIONS AND WARNINGS)
SORIATANE is contraindicated in patients with severely impaired liver or kidney function and in patients with chronic abnormally elevated blood lipid values (see boxed WARNINGS: Hepatotoxicity, WARNINGS: Lipids and Possible Cardiovascular Effects, and PRECAUTIONS).
An increased risk of hepatitis has been reported to result from combined use of methotrexate and etretinate. Consequently, the combination of methotrexate with SORIATANE is also contraindicated (see PRECAUTIONS and DRUG INTERACTIONS).
Since both SORIATANE and tetracyclines can cause increased intracranial pressure, their combined use is contraindicated (see WARNINGS: Pseudotumor Cerebri).
SORIATANE is contraindicated in cases of hypersensitivity (e.g., angioedema, urticaria) to the preparation (acitretin or excipients) or to other retinoids.
Undesirable effects
Hypervitaminosis A produces a wide spectrum of signs and symptoms primarily of the mucocutaneous, musculoskeletal, hepatic, neuropsychiatric, and central nervous systems. Many of the clinical adverse reactions reported to date with administration of SORIATANE resemble those of the hypervitaminosis A syndrome.
Adverse Events/Postmarketing ReportsIn addition to the events listed in the tables for the clinical trials, the following adverse events have been identified during postapproval use of SORIATANE. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiovascular: Acute myocardial infarction, thromboembolism (see WARNINGS), stroke.
Immune System Disorders: Hypersensitivity, including angioedema and urticaria (see CONTRAINDICATIONS).
Nervous System: Myopathy with peripheral neuropathy has been reported during therapy with SORIATANE. Both conditions improved with discontinuation of the drug.
Psychiatric: Aggressive feelings and/or suicidal thoughts have been reported. These events, including self-injurious behavior, have been reported in patients taking other systemically administered retinoids, as well as in patients taking SORIATANE. Since other factors may have contributed to these events, it is not known if they are related to SORIATANE (see PRECAUTIONS).
Reproductive: Vulvo-vaginitis due to Candida albicans.
Skin and Appendages: Thinning of the skin, skin fragility, and scaling may occur all over the body, particularly on the palms and soles; nail fragility is frequently observed. Madarosis and exfoliative dermatitis/erythroderma have been reported (see WARNINGS).
Vascular Disorders: Capillary leak syndrome (see WARNINGS).
Clinical TrialsDuring clinical trials with SORIATANE, 513 of 525 (98%) subjects reported a total of 3,545 adverse events. One-hundred sixteen subjects (22%) left trials prematurely, primarily because of adverse experiences involving the mucous membranes and skin. Three subjects died. Two of the deaths were not drug-related (pancreatic adenocarcinoma and lung cancer); the other subject died of an acute myocardial infarction, considered remotely related to drug therapy. In clinical trials, SORIATANE was associated with elevations in liver function test results or triglyceride levels and hepatitis.
The tables below list by body system and frequency the adverse events reported during clinical trials of 525 subjects with psoriasis.
Table 3. Adverse Events Frequently Reported during Clinical Trials Percent of Subjects Reporting (N = 525)
| Body System | >75% | 50% to 75% | 25% to 50% | 10% to 25% |
| CNS | Rigors | |||
| Eye Disorders | Xerophthalmia | |||
| Mucous Membranes | Cheilitis | Rhinitis | Dry mouth Epistaxis |
|
| Musculoskeletal | Arthralgia Spinal hyperostosis (progression of existing lesions) |
|||
| Skin and Appendages | Alopecia Skin peeling | Dry skin Nail disorder Pruritus |
Erythematous rash Hyperesthesia Paresthesia Paronychia Skin atrophy Sticky skin |
Table 4. Adverse Events Less Frequently Reported during Clinical Trials (Some of Which
May Bear No Relationship to Therapy)
Percent of Subjects Reporting (N = 525)
| Body System | 1% to 10% | <1% | ||
| Body as a Whole | Anorexia Edema Fatigue Hot flashes Increased appetite |
Alcohol intolerance Dizziness Fever Influenza-like symptoms |
Malaise Moniliasis Muscle weakness Weight increase |
|
| Cardiovascular | Flushing | Chest pain Cyanosis Increased bleeding time |
Intermittent claudication Peripheral ischemia |
|
| CNS (also see Psychiatric) | Headache Pain | Abnormal gait Migraine Neuritis |
Pseudotumor cerebri (intracranial hypertension) | |
| Eye Disorders | Abnormal/ blurred vision Blepharitis Conjunctivitis/ irritation Corneal epithelial abnormality |
Decreased night vision/night blindness Eye abnormality Eye pain Photophobia |
Abnormal lacrimation Chalazion Conjunctival hemorrhage Corneal ulceration Diplopia Ectropion |
Itchy eyes and lids Papilledema Recurrent sties Subepithelial corneal lesions |
| Gastrointestinal | Abdominal pain Diarrhea Nausea Tongue disorder |
Constipation Dyspepsia Esophagitis Gastritis Gastroenteritis |
Glossitis Hemorrhoids Melena Tenesmus Tongue ulceration |
|
| Liver and Biliary | Hepatic function abnormal Hepatitis Jaundice |
|||
| Mucous Membranes |
Gingival bleeding Gingivitis Increased saliva |
Stomatitis Thirst Ulcerative stomatitis |
Altered saliva Anal disorder Gum hyperplasia |
Hemorrhage Pharyngitis |
| Musculoskeletal | Arthritis Arthrosis Back pain Hypertonia Myalgia |
Osteodynia Peripheral joint hyperostosis (progression of existing lesions) |
Bone disorder Olecranon bursitis Spinal hyperostosis (new lesions) Tendonitis |
|
| Psychiatric | Depression Insomnia Somnolence |
Anxiety Dysphonia Libido decreased Nervousness |
||
| Reproductive | Atrophic vaginitis Leukorrhea |
|||
| Respiratory | Sinusitis | Coughing Increased sputum Laryngitis |
||
| Skin and Appendages | Abnormal skin odor Abnormal hair texture Bullous eruption Cold/clammy skin Dermatitis Increased sweating Infection |
Psoriasiform rash Purpura Pyogenic granuloma Rash Seborrhea Skin fissures Skin ulceration Sunburn |
Acne Breast pain Cyst Eczema Fungal infection Furunculosis Hair discoloration Herpes simplex Hyperkeratosis Hypertrichosis Hypoesthesia Impaired healing Otitis media |
Otitis externa Photosensitivity reaction Psoriasis aggravated Scleroderma Skin nodule Skin hypertrophy Skin disorder Skin irritation Sweat gland disorder Urticaria Verrucae |
| Special Senses/ Other | Earache Taste perversion Tinnitus |
Ceruminosis Deafness Taste loss |
||
| Urinary | Abnormal urine Dysuria Penis disorder |
Therapy with SORIATANE induces changes in liver function tests in a significant number of patients. Elevations of AST (SGOT), ALT (SGPT) or LDH were experienced by approximately 1 in 3 subjects treated with SORIATANE. In most subjects, elevations were slight to moderate and returned to normal either during continuation of therapy or after cessation of treatment. In subjects receiving SORIATANE during clinical trials, 66% and 33% experienced elevation in triglycerides and cholesterol, respectively. Decreased high density lipoproteins (HDL) occurred in 40% (see WARNINGS). Transient, usually reversible elevations of alkaline phosphatase have been observed.
Table 5 lists the laboratory abnormalities reported during clinical trials.
Table 5. Abnormal Laboratory Test Results Reported during Clinical Trials Percent of Subjects Reporting
| Body System | 50% to 75% | 25% to 50% | 10% to 25% | 1% to 10% |
| Electrolytes | Increased:
|
Decreased:
|
||
| Hematologic | Increased:
|
Decreased:
|
Increased:
|
|
| Hepatic | Increased:
|
Increased:
|
Increased:
|
|
| Miscellaneous | Increased:
|
Increased:
|
Decreased:
|
Increased and decreased:
|
| Renal | Increased:
|
Increased:
|
||
| Urinary | WBC in urine | Acetonuria Hematuria RBC in urine | Glycosuria Proteinuria |
Therapeutic indications
SORIATANE is indicated for the treatment of severe psoriasis in adults. Because of significant adverse effects associated with its use, SORIATANE should be prescribed only by those knowledgeable in the systemic use of retinoids. In females of reproductive potential, SORIATANE should be reserved for non-pregnant patients who are unresponsive to other therapies or whose clinical condition contraindicates the use of other treatments (see boxed CONTRAINDICATIONS AND WARNINGS - SORIATANE can cause severe birth defects).
Most patients experience relapse of psoriasis after discontinuing therapy. Subsequent courses, when clinically indicated, have produced efficacy results similar to the initial course of therapy.
Pharmacokinetic properties
). GlyburideIn a trial of 7 healthy male volunteers, acitretin treatment potentiated the blood glucose-lowering effect of glyburide (a sulfonylurea similar to chlorpropamide) in 3 of the 7 subjects. Repeating the trial with 6 healthy male volunteers in the absence of glyburide did not detect an effect of acitretin on glucose tolerance. Careful supervision of diabetic patients under treatment with SORIATANE is recommended (see CLINICAL PHARMACOLOGY: Pharmacokinetics and DOSAGE AND ADMINISTRATION).
Hormonal ContraceptivesIt has not been established if there is a pharmacokinetic interaction between acitretin and combined oral contraceptives. However, it has been established that acitretin interferes with the contraceptive effect of microdosed progestin “minipill” preparations. Microdosed “minipill” progestin preparations are not recommended for use with SORIATANE (see CLINICAL PHARMACOLOGY: Pharmacokinetic Drug Interactions). It is not known whether other progestin-only contraceptives, such as implants and injectables, are adequate methods of contraception during acitretin therapy.
MethotrexateAn increased risk of hepatitis has been reported to result from combined use of methotrexate and etretinate. Consequently, the combination of methotrexate with acitretin is also contraindicated (see CONTRAINDICATIONS).
PhenytoinIf acitretin is given concurrently with phenytoin, the protein binding of phenytoin may be reduced.
TetracyclinesSince both acitretin and tetracyclines can cause increased intracranial pressure, their combined use is contraindicated (see CONTRAINDICATIONS and WARNINGS: Pseudotumor Cerebri).
Vitamin A And Oral RetinoidsConcomitant administration of vitamin A and/or other oral retinoids with acitretin must be avoided because of the risk of hypervitaminosis A.
OtherThere appears to be no pharmacokinetic interaction between acitretin and cimetidine, digoxin, or glyburide. Investigations into the effect of acitretin on the protein binding of anticoagulants of the coumarin type (warfarin) revealed no interaction.
Warnings & Precautions WARNINGS(See also boxed CONTRAINDICATIONS AND WARNINGS)
HepatotoxicityOf the 525 subjects treated in US clinical trials, 2 had clinical jaundice with elevated serum bilirubin and transaminases considered related to treatment with SORIATANE. Liver function test results in these subjects returned to normal after SORIATANE was discontinued. Two of the 1,289 subjects treated in European clinical trials developed biopsy-confirmed toxic hepatitis. A second biopsy in one of these subjects revealed nodule formation suggestive of cirrhosis. One subject in a Canadian clinical trial of 63 subjects developed a 3-fold increase of transaminases. A liver biopsy of this subject showed mild lobular disarray, multifocal hepatocyte loss, and mild triaditis of the portal tracts compatible with acute reversible hepatic injury. The subject's transaminase levels returned to normal 2 months after SORIATANE was discontinued.
The potential of therapy with SORIATANE to induce hepatotoxicity was prospectively evaluated using liver biopsies in an open-label trial of 128 subjects. Pretreatment and posttreatment biopsies were available for 87 subjects. A comparison of liver biopsy findings before and after therapy revealed 49 (58%) subjects showed no change, 21 (25%) improved, and 14 (17%) subjects had a worsening of their liver biopsy status. For 6 subjects, the classification changed from class 0 (no pathology) to class I (normal fatty infiltration; nuclear variability and portal inflammation; both mild); for 7 subjects, the change was from class I to class II (fatty infiltration, nuclear variability, portal inflammation, and focal necrosis; all moderate to severe); and for 1 subject, the change was from class II to class IIIb (fibrosis, moderate to severe). No correlation could be found between liver function test result abnormalities and the change in liver biopsy status, and no cumulative dose relationship was found.
Elevations of AST (SGOT), ALT (SGPT), GGT (GGTP), or LDH have occurred in approximately 1 in 3 subjects treated with SORIATANE. Of the 525 subjects treated in clinical trials in the US, treatment was discontinued in 20 (3.8%) due to elevated liver function test results. If hepatotoxicity is suspected during treatment with SORIATANE, the drug should be discontinued and the etiology further investigated.
Ten of 652 subjects treated in US clinical trials of etretinate, of which acitretin is the active metabolite, had clinical or histologic hepatitis considered to be possibly or probably related to etretinate treatment.
There have been reports of hepatitis-related deaths worldwide; a few of these subjects had received etretinate for a month or less before presenting with hepatic symptoms or signs.
Skeletal AbnormalitiesIn adults receiving long-term treatment with SORIATANE, appropriate examinations should be periodically performed in view of possible ossification abnormalities (see ADVERSE REACTIONS). Because the frequency and severity of iatrogenic bony abnormality in adults is low, periodic radiography is only warranted in the presence of symptoms or long-term use of SORIATANE. If such disorders arise, the continuation of therapy should be discussed with the patient on the basis of a careful risk/benefit analysis. In clinical trials with SORIATANE, subjects were prospectively evaluated for evidence of development or change in bony abnormalities of the vertebral column, knees, and ankles.
Of 380 subjects treated with SORIATANE, 15% had preexisting abnormalities of the spine which showed new changes or progression of preexisting findings. Changes included degenerative spurs, anterior bridging of spinal vertebrae, diffuse idiopathic skeletal hyperostosis, ligament calcification, and narrowing and destruction of a cervical disc space. De novo changes (formation of small spurs) were seen in 3 subjects after 1½ to 2½ years.
Six of 128 subjects treated with SORIATANE showed abnormalities in the knees and ankles before treatment that progressed during treatment. In 5, these changes involved the formation of additional spurs or enlargement of existing spurs. The sixth subject had degenerative joint disease which worsened. No subjects developed spurs de novo. Clinical complaints did not predict radiographic changes.
Lipids And Possible Cardiovascular EffectsBlood lipid determinations should be performed before SORIATANE is administered and again at intervals of 1 to 2 weeks until the lipid response to the drug is established, usually within 4 to 8 weeks. In subjects receiving SORIATANE during clinical trials, 66% and 33% experienced elevation in triglycerides and cholesterol, respectively. Decreased high density lipoproteins (HDL) occurred in 40% of subjects. These effects of SORIATANE were generally reversible upon cessation of therapy.
Subjects with an increased tendency to develop hypertriglyceridemia included those with disturbances of lipid metabolism, diabetes mellitus, obesity, increased alcohol intake, or a familial history of these conditions. Because of the risk of hypertriglyceridemia, serum lipids must be more closely monitored in high-risk patients and during long-term treatment.
Hypertriglyceridemia and lowered HDL may increase a patient’s cardiovascular risk status. Although no causal relationship has been established, there have been postmarketing reports of acute myocardial infarction or thromboembolic events in patients on therapy with SORIATANE. In addition, elevation of serum triglycerides to greater than 800 mg per dL has been associated with fatal fulminant pancreatitis. Therefore, dietary modifications, reduction in dose of SORIATANE, or drug therapy should be employed to control significant elevations of triglycerides. If, despite these measures, hypertriglyceridemia and low HDL levels persist, the discontinuation of SORIATANE should be considered.
Ophthalmologic EffectsThe eyes and vision of 329 subjects treated with SORIATANE were examined by ophthalmologists. The findings included dry eyes (23%), irritation of eyes (9%), and brow and lash loss (5%). The following were reported in less than 5% of subjects: Bell’s palsy, blepharitis and/or crusting of lids, blurred vision, conjunctivitis, corneal epithelial abnormality, cortical cataract, decreased night vision, diplopia, itchy eyes or eyelids, nuclear cataract, pannus, papilledema, photophobia, posterior subcapsular cataract, recurrent sties, and subepithelial corneal lesions.
Any patient treated with SORIATANE who is experiencing visual difficulties should discontinue the drug and undergo ophthalmologic evaluation.
PancreatitisLipid elevations occur in 25% to 50% of subjects treated with SORIATANE. Triglyceride increases sufficient to be associated with pancreatitis are much less common, although fatal fulminant pancreatitis has been reported. There have been rare reports of pancreatitis during therapy with SORIATANE in the absence of hypertriglyceridemia.
Pseudotumor CerebriSORIATANE and other retinoids administered orally have been associated with cases of pseudotumor cerebri (benign intracranial hypertension). Some of these events involved concomitant use of isotretinoin and tetracyclines. However, the event seen in a single patient receiving SORIATANE was not associated with tetracycline use. Early signs and symptoms include papilledema, headache, nausea and vomiting, and visual disturbances. Patients with these signs and symptoms should be examined for papilledema and, if present, should discontinue SORIATANE immediately and be referred for neurological evaluation and care. Since both SORIATANE and tetracyclines can cause increased intracranial pressure, their combined use is contraindicated (see CONTRAINDICATIONS).
Capillary Leak SyndromeCapillary leak syndrome, a potential manifestation of retinoic acid syndrome, has been reported in patients receiving SORIATANE. Features of this syndrome may include localized or generalized edema with secondary weight gain, fever, and hypotension. Rhabdomyolysis and myalgias have been reported in association with capillary leak syndrome, and laboratory tests may reveal neutrophilia, hypoalbuminemia, and an elevated hematocrit. Discontinue SORIATANE if capillary leak syndrome develops during therapy.
Exfoliative Dermatitis/ErythrodermaExfoliative dermatitis/erythroderma has been reported in patients receiving SORIATANE. Discontinue SORIATANE if exfoliative dermatitis/erythroderma occurs during therapy.
PRECAUTIONSA description of the Do Your P.A.R.T. materials is provided below. The main goals of the materials are to explain the program requirements, to reinforce the educational messages, and to assess program effectiveness.
The Do Your P.A.R.T. booklet includes:
- The Do Your P.A.R.T. Patient Brochure: information on the program requirements, risks of acitretin, and the types of contraceptive methods
- The Contraception Counseling Referral Form for female patients who want to receive free contraception counseling reimbursed by the manufacturer
- The Patient Agreement/Informed Consent for Female Patients form
Date of revision of the text
Sep 2017Qualitative and quantitative composition
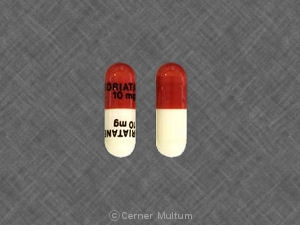
Brown and white capsules, 10 mg, imprinted “A-10 mg”; bottles of 30 (NDC 0145-0090-25).
Rich yellow capsules, 17.5 mg, imprinted “A-17.5 mg”; bottles of 30 (NDC 0145-3817-03).
Brown and yellow capsules, 25 mg, imprinted “A-25 mg”; bottles of 30 (NDC 0145-009125).
Store between 15° and 25°C (59° and 77°F). Protect from light. Avoid exposure to high temperatures and humidity after the bottle is opened.
Manufactured for: Stiefel Laboratories, Inc., Research Triangle Park, NC 27709. Revised: Sep 2017
Special warnings and precautions for use
WARNINGS(See also boxed CONTRAINDICATIONS AND WARNINGS)
HepatotoxicityOf the 525 subjects treated in US clinical trials, 2 had clinical jaundice with elevated serum bilirubin and transaminases considered related to treatment with SORIATANE. Liver function test results in these subjects returned to normal after SORIATANE was discontinued. Two of the 1,289 subjects treated in European clinical trials developed biopsy-confirmed toxic hepatitis. A second biopsy in one of these subjects revealed nodule formation suggestive of cirrhosis. One subject in a Canadian clinical trial of 63 subjects developed a 3-fold increase of transaminases. A liver biopsy of this subject showed mild lobular disarray, multifocal hepatocyte loss, and mild triaditis of the portal tracts compatible with acute reversible hepatic injury. The subject's transaminase levels returned to normal 2 months after SORIATANE was discontinued.
The potential of therapy with SORIATANE to induce hepatotoxicity was prospectively evaluated using liver biopsies in an open-label trial of 128 subjects. Pretreatment and posttreatment biopsies were available for 87 subjects. A comparison of liver biopsy findings before and after therapy revealed 49 (58%) subjects showed no change, 21 (25%) improved, and 14 (17%) subjects had a worsening of their liver biopsy status. For 6 subjects, the classification changed from class 0 (no pathology) to class I (normal fatty infiltration; nuclear variability and portal inflammation; both mild); for 7 subjects, the change was from class I to class II (fatty infiltration, nuclear variability, portal inflammation, and focal necrosis; all moderate to severe); and for 1 subject, the change was from class II to class IIIb (fibrosis, moderate to severe). No correlation could be found between liver function test result abnormalities and the change in liver biopsy status, and no cumulative dose relationship was found.
Elevations of AST (SGOT), ALT (SGPT), GGT (GGTP), or LDH have occurred in approximately 1 in 3 subjects treated with SORIATANE. Of the 525 subjects treated in clinical trials in the US, treatment was discontinued in 20 (3.8%) due to elevated liver function test results. If hepatotoxicity is suspected during treatment with SORIATANE, the drug should be discontinued and the etiology further investigated.
Ten of 652 subjects treated in US clinical trials of etretinate, of which acitretin is the active metabolite, had clinical or histologic hepatitis considered to be possibly or probably related to etretinate treatment.
There have been reports of hepatitis-related deaths worldwide; a few of these subjects had received etretinate for a month or less before presenting with hepatic symptoms or signs.
Skeletal AbnormalitiesIn adults receiving long-term treatment with SORIATANE, appropriate examinations should be periodically performed in view of possible ossification abnormalities (see ADVERSE REACTIONS). Because the frequency and severity of iatrogenic bony abnormality in adults is low, periodic radiography is only warranted in the presence of symptoms or long-term use of SORIATANE. If such disorders arise, the continuation of therapy should be discussed with the patient on the basis of a careful risk/benefit analysis. In clinical trials with SORIATANE, subjects were prospectively evaluated for evidence of development or change in bony abnormalities of the vertebral column, knees, and ankles.
Of 380 subjects treated with SORIATANE, 15% had preexisting abnormalities of the spine which showed new changes or progression of preexisting findings. Changes included degenerative spurs, anterior bridging of spinal vertebrae, diffuse idiopathic skeletal hyperostosis, ligament calcification, and narrowing and destruction of a cervical disc space. De novo changes (formation of small spurs) were seen in 3 subjects after 1½ to 2½ years.
Six of 128 subjects treated with SORIATANE showed abnormalities in the knees and ankles before treatment that progressed during treatment. In 5, these changes involved the formation of additional spurs or enlargement of existing spurs. The sixth subject had degenerative joint disease which worsened. No subjects developed spurs de novo. Clinical complaints did not predict radiographic changes.
Lipids And Possible Cardiovascular EffectsBlood lipid determinations should be performed before SORIATANE is administered and again at intervals of 1 to 2 weeks until the lipid response to the drug is established, usually within 4 to 8 weeks. In subjects receiving SORIATANE during clinical trials, 66% and 33% experienced elevation in triglycerides and cholesterol, respectively. Decreased high density lipoproteins (HDL) occurred in 40% of subjects. These effects of SORIATANE were generally reversible upon cessation of therapy.
Subjects with an increased tendency to develop hypertriglyceridemia included those with disturbances of lipid metabolism, diabetes mellitus, obesity, increased alcohol intake, or a familial history of these conditions. Because of the risk of hypertriglyceridemia, serum lipids must be more closely monitored in high-risk patients and during long-term treatment.
Hypertriglyceridemia and lowered HDL may increase a patient’s cardiovascular risk status. Although no causal relationship has been established, there have been postmarketing reports of acute myocardial infarction or thromboembolic events in patients on therapy with SORIATANE. In addition, elevation of serum triglycerides to greater than 800 mg per dL has been associated with fatal fulminant pancreatitis. Therefore, dietary modifications, reduction in dose of SORIATANE, or drug therapy should be employed to control significant elevations of triglycerides. If, despite these measures, hypertriglyceridemia and low HDL levels persist, the discontinuation of SORIATANE should be considered.
Ophthalmologic EffectsThe eyes and vision of 329 subjects treated with SORIATANE were examined by ophthalmologists. The findings included dry eyes (23%), irritation of eyes (9%), and brow and lash loss (5%). The following were reported in less than 5% of subjects: Bell’s palsy, blepharitis and/or crusting of lids, blurred vision, conjunctivitis, corneal epithelial abnormality, cortical cataract, decreased night vision, diplopia, itchy eyes or eyelids, nuclear cataract, pannus, papilledema, photophobia, posterior subcapsular cataract, recurrent sties, and subepithelial corneal lesions.
Any patient treated with SORIATANE who is experiencing visual difficulties should discontinue the drug and undergo ophthalmologic evaluation.
PancreatitisLipid elevations occur in 25% to 50% of subjects treated with SORIATANE. Triglyceride increases sufficient to be associated with pancreatitis are much less common, although fatal fulminant pancreatitis has been reported. There have been rare reports of pancreatitis during therapy with SORIATANE in the absence of hypertriglyceridemia.
Pseudotumor CerebriSORIATANE and other retinoids administered orally have been associated with cases of pseudotumor cerebri (benign intracranial hypertension). Some of these events involved concomitant use of isotretinoin and tetracyclines. However, the event seen in a single patient receiving SORIATANE was not associated with tetracycline use. Early signs and symptoms include papilledema, headache, nausea and vomiting, and visual disturbances. Patients with these signs and symptoms should be examined for papilledema and, if present, should discontinue SORIATANE immediately and be referred for neurological evaluation and care. Since both SORIATANE and tetracyclines can cause increased intracranial pressure, their combined use is contraindicated (see CONTRAINDICATIONS).
Capillary Leak SyndromeCapillary leak syndrome, a potential manifestation of retinoic acid syndrome, has been reported in patients receiving SORIATANE. Features of this syndrome may include localized or generalized edema with secondary weight gain, fever, and hypotension. Rhabdomyolysis and myalgias have been reported in association with capillary leak syndrome, and laboratory tests may reveal neutrophilia, hypoalbuminemia, and an elevated hematocrit. Discontinue SORIATANE if capillary leak syndrome develops during therapy.
Exfoliative Dermatitis/ErythrodermaExfoliative dermatitis/erythroderma has been reported in patients receiving SORIATANE. Discontinue SORIATANE if exfoliative dermatitis/erythroderma occurs during therapy.
PRECAUTIONSA description of the Do Your P.A.R.T. materials is provided below. The main goals of the materials are to explain the program requirements, to reinforce the educational messages, and to assess program effectiveness.
The Do Your P.A.R.T. booklet includes:
- The Do Your P.A.R.T. Patient Brochure: information on the program requirements, risks of acitretin, and the types of contraceptive methods
- The Contraception Counseling Referral Form for female patients who want to receive free contraception counseling reimbursed by the manufacturer
- The Patient Agreement/Informed Consent for Female Patients form
- Medication Guide
The Do Your P.A.R.T. program also includes a voluntary patient survey for women of childbearing potential to assess the effectiveness of the SORIATANE Pregnancy Prevention Program Do Your P.A.R.T. Do Your P.A.R.T. Program materials are available at www.soriatane.com or may be requested by calling 1-888-784-3335 (1-888-STIEFEL).
Information For Patients(See PATIENT INFORMATION for all patients and Patient Agreement/Informed Consent for Female Patients at end of professional labeling).
Patients should be instructed to read the Medication Guide supplied as required by law when SORIATANE is dispensed.
Females Of Reproductive PotentialSORIATANE can cause severe birth defects. Female patients must not be pregnant when therapy with SORIATANE is initiated, they must not become pregnant while taking SORIATANE and for at least 3 years after stopping SORIATANE, so that the drug can be eliminated to below a blood concentration that would be associated with an increased incidence of birth defects. Because this threshold has not been established for acitretin in humans and because elimination rates vary among patients, the duration of posttherapy contraception to achieve adequate elimination cannot be calculated precisely (see boxed CONTRAINDICATIONS AND WARNINGS).
Females of reproductive potential should also be advised that they must not ingest beverages or products containing ethanol while taking SORIATANE and for 2 months after SORIATANE has been discontinued. This allows for elimination of the acitretin which can be converted to etretinate in the presence of alcohol.
Female patients should be advised that any method of birth control can fail, including tubal ligation, and that microdosed progestin “minipill” preparations are not recommended for use with SORIATANE (see CLINICAL PHARMACOLOGY: Pharmacokinetic Drug Interactions). Data from one patient who received a very low-dosed progestin contraceptive (levonorgestrel 0.03 mg) had a significant increase of the progesterone level after 3 menstrual cycles during acitretin treatment.2
Female patients should be advised to contact their physician, women’s health centers, pharmacies, or hospital emergency rooms for information about how to obtain Emergency Contraception if sexual intercourse occurs without using 2 effective forms of contraception simultaneously. A 24-hour, toll-free number (1-800-739-6700) is also available for patients to receive automated birth control and emergency contraception information.
Female patients should sign a consent form prior to beginning therapy with SORIATANE (see boxed CONTRAINDICATIONS AND WARNINGS).
Nursing MothersStudies on lactating rats have shown that etretinate is excreted in the milk. There is one prospective case report where acitretin is reported to be excreted in human milk. Therefore, nursing mothers should not receive SORIATANE prior to or during nursing because of the potential for serious adverse reactions in nursing infants.
All PatientsDepression and/or other psychiatric symptoms such as aggressive feelings or thoughts of self-harm have been reported. These events, including self-injurious behavior, have been reported in patients taking other systemically administered retinoids, as well as in patients taking SORIATANE. Since other factors may have contributed to these events, it is not known if they are related to SORIATANE. Patients should be counseled to stop taking SORIATANE and notify their prescriber immediately if they experience psychiatric symptoms.
Patients should be advised that a transient worsening of psoriasis is sometimes seen during the initial treatment period. Patients should be advised that they may have to wait 2 to 3 months before they get the full benefit of SORIATANE, although some patients may achieve significant improvements within the first 8 weeks of treatment as demonstrated in clinical trials.
Decreased night vision has been reported during therapy with SORIATANE. Patients should be advised of this potential problem and warned to be cautious when driving or operating any vehicle at night. Visual problems should be carefully monitored (see WARNINGS and ADVERSE REACTIONS). Patients should be advised that they may experience decreased tolerance to contact lenses during the treatment period and sometimes after treatment has stopped.
Patients should not donate blood during and for at least 3 years following therapy because SORIATANE can cause birth defects and women of childbearing potential must not receive blood from patients being treated with SORIATANE.
Because of the relationship of SORIATANE to vitamin A, patients should be advised against taking vitamin A supplements in excess of minimum recommended daily allowances to avoid possible additive toxic effects.
Patients should avoid the use of sun lamps and excessive exposure to sunlight (non-medical UV exposure) because the effects of UV light are enhanced by retinoids.
Patients should be advised that they must not give their SORIATANE to any other person.
For PrescribersSORIATANE has not been studied in and is not indicated for treatment of acne.
PhototherapySignificantly lower doses of phototherapy are required when SORIATANE is used because effects on the stratum corneum induced by SORIATANE can increase the risk of erythema (burning) (see DOSAGE AND ADMINISTRATION).
REFERENCES
2. Maier H, Honigsmann H: Concentration of etretinate in plasma and subcutaneous fat after long-term acitretin. Lancet 348:1107, 1996.
4. Sigg C, et al.: Andrological investigations in patients treated with etretin. Dermatologica 175:48-49, 1987.
5.Parsch EM, et al.: Andrological investigation in men treated with acitretin (Ro 10-1670). Andrologia 22:479-482, 1990.
6. Kadar L, et al.: Spermatological investigations in psoriatic patients treated with acitretin. In: Pharmacology of Retinoids in the Skin; Reichert U. et al., ed, KARGER, Basel, vol. 3, pp 253-254, 1988.
Dosage (Posology) and method of administration
There is intersubject variation in the pharmacokinetics, clinical efficacy, and incidence of side effects with SORIATANE. A number of the more common side effects are dose-related. Individualization of dosage is required to achieve sufficient therapeutic response while minimizing side effects. Therapy with SORIATANE should be initiated at 25 to 50 mg per day, given as a single dose with the main meal. Maintenance doses of 25 to 50 mg per day may be given dependent upon an individual patient’s response to initial treatment. Relapses may be treated as outlined for initial therapy.
When SORIATANE is used with phototherapy, the prescriber should decrease the phototherapy dose, dependent on the patient’s individual response (see PRECAUTIONS: General).
Females who have taken TEGISON (etretinate) must continue to follow the contraceptive recommendations for TEGISON. TEGISON is no longer marketed in the US; for information, call Stiefel at 1-888-784-3335 (1-888-STIEFEL).
Information For PharmacistsSORIATANE must only be dispensed in no more than a monthly supply. A SORIATANE Medication Guide must be given to the patient each time SORIATANE is dispensed, as required by law.
Interaction with other medicinal products and other forms of interaction
SIDE EFFECTSHypervitaminosis A produces a wide spectrum of signs and symptoms primarily of the mucocutaneous, musculoskeletal, hepatic, neuropsychiatric, and central nervous systems. Many of the clinical adverse reactions reported to date with administration of SORIATANE resemble those of the hypervitaminosis A syndrome.
Adverse Events/Postmarketing ReportsIn addition to the events listed in the tables for the clinical trials, the following adverse events have been identified during postapproval use of SORIATANE. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiovascular: Acute myocardial infarction, thromboembolism (see WARNINGS), stroke.
Immune System Disorders: Hypersensitivity, including angioedema and urticaria (see CONTRAINDICATIONS).
Nervous System: Myopathy with peripheral neuropathy has been reported during therapy with SORIATANE. Both conditions improved with discontinuation of the drug.
Psychiatric: Aggressive feelings and/or suicidal thoughts have been reported. These events, including self-injurious behavior, have been reported in patients taking other systemically administered retinoids, as well as in patients taking SORIATANE. Since other factors may have contributed to these events, it is not known if they are related to SORIATANE (see PRECAUTIONS).
Reproductive: Vulvo-vaginitis due to Candida albicans.
Skin and Appendages: Thinning of the skin, skin fragility, and scaling may occur all over the body, particularly on the palms and soles; nail fragility is frequently observed. Madarosis and exfoliative dermatitis/erythroderma have been reported (see WARNINGS).
Vascular Disorders: Capillary leak syndrome (see WARNINGS).
Clinical TrialsDuring clinical trials with SORIATANE, 513 of 525 (98%) subjects reported a total of 3,545 adverse events. One-hundred sixteen subjects (22%) left trials prematurely, primarily because of adverse experiences involving the mucous membranes and skin. Three subjects died. Two of the deaths were not drug-related (pancreatic adenocarcinoma and lung cancer); the other subject died of an acute myocardial infarction, considered remotely related to drug therapy. In clinical trials, SORIATANE was associated with elevations in liver function test results or triglyceride levels and hepatitis.
The tables below list by body system and frequency the adverse events reported during clinical trials of 525 subjects with psoriasis.
Table 3. Adverse Events Frequently Reported during Clinical Trials Percent of Subjects Reporting (N = 525)
| Body System | >75% | 50% to 75% | 25% to 50% | 10% to 25% |
| CNS | Rigors | |||
| Eye Disorders | Xerophthalmia | |||
| Mucous Membranes | Cheilitis | Rhinitis | Dry mouth Epistaxis |
|
| Musculoskeletal | Arthralgia Spinal hyperostosis (progression of existing lesions) |
|||
| Skin and Appendages | Alopecia Skin peeling | Dry skin Nail disorder Pruritus |
Erythematous rash Hyperesthesia Paresthesia Paronychia Skin atrophy Sticky skin |
Table 4. Adverse Events Less Frequently Reported during Clinical Trials (Some of Which
May Bear No Relationship to Therapy)
Percent of Subjects Reporting (N = 525)
| Body System | 1% to 10% | <1% | ||
| Body as a Whole | Anorexia Edema Fatigue Hot flashes Increased appetite |
Alcohol intolerance Dizziness Fever Influenza-like symptoms |
Malaise Moniliasis Muscle weakness Weight increase |
|
| Cardiovascular | Flushing | Chest pain Cyanosis Increased bleeding time |
Intermittent claudication Peripheral ischemia |
|
| CNS (also see Psychiatric) | Headache Pain | Abnormal gait Migraine Neuritis |
Pseudotumor cerebri (intracranial hypertension) | |
| Eye Disorders | Abnormal/ blurred vision Blepharitis Conjunctivitis/ irritation Corneal epithelial abnormality |
Decreased night vision/night blindness Eye abnormality Eye pain Photophobia |
Abnormal lacrimation Chalazion Conjunctival hemorrhage Corneal ulceration Diplopia Ectropion |
Itchy eyes and lids Papilledema Recurrent sties Subepithelial corneal lesions |
| Gastrointestinal | Abdominal pain Diarrhea Nausea Tongue disorder |
Constipation Dyspepsia Esophagitis Gastritis Gastroenteritis |
Glossitis Hemorrhoids Melena Tenesmus Tongue ulceration |
|
| Liver and Biliary | Hepatic function abnormal Hepatitis Jaundice |
|||
| Mucous Membranes |
Gingival bleeding Gingivitis Increased saliva |
Stomatitis Thirst Ulcerative stomatitis |
Altered saliva Anal disorder Gum hyperplasia |
Hemorrhage Pharyngitis |
| Musculoskeletal | Arthritis Arthrosis Back pain Hypertonia Myalgia |
Osteodynia Peripheral joint hyperostosis (progression of existing lesions) |
Bone disorder Olecranon bursitis Spinal hyperostosis (new lesions) Tendonitis |
|
| Psychiatric | Depression Insomnia Somnolence |
Anxiety Dysphonia Libido decreased Nervousness |
||
| Reproductive | Atrophic vaginitis Leukorrhea |
|||
| Respiratory | Sinusitis | Coughing Increased sputum Laryngitis |
||
| Skin and Appendages | Abnormal skin odor Abnormal hair texture Bullous eruption Cold/clammy skin Dermatitis Increased sweating Infection |
Psoriasiform rash Purpura Pyogenic granuloma Rash Seborrhea Skin fissures Skin ulceration Sunburn |
Acne Breast pain Cyst Eczema Fungal infection Furunculosis Hair discoloration Herpes simplex Hyperkeratosis Hypertrichosis Hypoesthesia Impaired healing Otitis media |
Otitis externa Photosensitivity reaction Psoriasis aggravated Scleroderma Skin nodule Skin hypertrophy Skin disorder Skin irritation Sweat gland disorder Urticaria Verrucae |
| Special Senses/ Other | Earache Taste perversion Tinnitus |
Ceruminosis Deafness Taste loss |
||
| Urinary | Abnormal urine Dysuria Penis disorder |
Therapy with SORIATANE induces changes in liver function tests in a significant number of patients. Elevations of AST (SGOT), ALT (SGPT) or LDH were experienced by approximately 1 in 3 subjects treated with SORIATANE. In most subjects, elevations were slight to moderate and returned to normal either during continuation of therapy or after cessation of treatment. In subjects receiving SORIATANE during clinical trials, 66% and 33% experienced elevation in triglycerides and cholesterol, respectively. Decreased high density lipoproteins (HDL) occurred in 40% (see WARNINGS). Transient, usually reversible elevations of alkaline phosphatase have been observed.
Table 5 lists the laboratory abnormalities reported during clinical trials.
Table 5. Abnormal Laboratory Test Results Reported during Clinical Trials Percent of Subjects Reporting
| Body System | 50% to 75% | 25% to 50% | 10% to 25% | 1% to 10% |
| Electrolytes | Increased:
|
Decreased:
|
||
| Hematologic | Increased:
|
Decreased:
|
Increased:
|
|
| Hepatic | Increased:
|
Increased:
|
Increased:
|
|
| Miscellaneous | Increased:
|
Increased:
|
Decreased:
|
Increased and decreased:
|
| Renal | Increased:
|
Increased:
|
||
| Urinary | WBC in urine | Acetonuria Hematuria RBC in urine | Glycosuria Proteinuria |
Clinical evidence has shown that etretinate can be formed with concurrent ingestion of acitretin and ethanol (see boxed CONTRAINDICATIONS AND WARNINGS and CONTRAINDICATIONS: Pharmacokinetics).
GlyburideIn a trial of 7 healthy male volunteers, acitretin treatment potentiated the blood glucose-lowering effect of glyburide (a sulfonylurea similar to chlorpropamide) in 3 of the 7 subjects. Repeating the trial with 6 healthy male volunteers in the absence of glyburide did not detect an effect of acitretin on glucose tolerance. Careful supervision of diabetic patients under treatment with SORIATANE is recommended (see CLINICAL PHARMACOLOGY: Pharmacokinetics and DOSAGE AND ADMINISTRATION).
Hormonal ContraceptivesIt has not been established if there is a pharmacokinetic interaction between acitretin and combined oral contraceptives. However, it has been established that acitretin interferes with the contraceptive effect of microdosed progestin “minipill” preparations. Microdosed “minipill” progestin preparations are not recommended for use with SORIATANE (see CLINICAL PHARMACOLOGY: Pharmacokinetic Drug Interactions). It is not known whether other progestin-only contraceptives, such as implants and injectables, are adequate methods of contraception during acitretin therapy.
MethotrexateAn increased risk of hepatitis has been reported to result from combined use of methotrexate and etretinate. Consequently, the combination of methotrexate with acitretin is also contraindicated (see CONTRAINDICATIONS).
PhenytoinIf acitretin is given concurrently with phenytoin, the protein binding of phenytoin may be reduced.
TetracyclinesSince both acitretin and tetracyclines can cause increased intracranial pressure, their combined use is contraindicated (see CONTRAINDICATIONS and WARNINGS: Pseudotumor Cerebri).
Vitamin A And Oral RetinoidsConcomitant administration of vitamin A and/or other oral retinoids with acitretin must be avoided because of the risk of hypervitaminosis A.
OtherThere appears to be no pharmacokinetic interaction between acitretin and cimetidine, digoxin, or glyburide. Investigations into the effect of acitretin on the protein binding of anticoagulants of the coumarin type (warfarin) revealed no interaction.