Naprosyn (oral)
Drugs with the same active ingredients
Overdose
Symptoms following acute NSAID overdosages have been typically limited to lethargy, drowsiness, nausea, vomiting, and epigastric pain, which have been generally reversible with supportive care. Gastrointestinal bleeding has occurred. Hypertension, acute renal failure, respiratory depression, and coma have occurred, but were rare.Because naproxen sodium may be rapidly absorbed, high and early blood levels should be anticipated. A few patients have experienced convulsions, but it is not clear whether or not these were drug-related. It is not known what dose of the drug would be life threatening..
Manage patients with symptomatic and supportive care following an NSAID overdosage. There are no specific antidotes. Consider emesis and/or activated charcoal (60 to 100 grams in adults, 1 to 2 grams per kg of body weight in pediatric patients) and/or osmotic cathartic in symptomatic patients seen within four hours of ingestion or in patients with a large overdosage (5 to 10 times the recommended dosage). Forced diuresis, alkalinization of urine, hemodialysis, or hemoperfusion may not be useful due to high protein binding.
For additional information about overdosage treatment contact a poison control center (1-800-222-1222).
Contraindications
NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS are contraindicated in the following patients:
- Known hypersensitivity (e.g., anaphylactic reactions and serious skin reactions) to naproxen or any components of the drug product
- History of asthma, urticaria, or other allergic-type reactions after taking aspirin or other NSAIDs. Severe, sometimes fatal, anaphylactic reactions to NSAIDs have been reported in such patients
- In the setting of coronary artery bypass graft (CABG) surgery
Undesirable effects
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Cardiovascular Thrombotic Events
- GI Bleeding, Ulceration, and Perforation
- Hepatotoxicity
- Hypertension
- Heart Failure and Edema
- Renal Toxicity and Hyperkalemia
- Anaphylactic Reactions
- Serious Skin Reactions
- Hematologic Toxicity
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse reactions reported in controlled clinical trials in 960 patients treated for rheumatoid arthritis or osteoarthritis are listed below. In general, reactions in patients treated chronically were reported 2 to 10 times more frequently than they were in short-term studies in the 962 patients treated for mild to moderate pain or for dysmenorrhea. The most frequent complaints reported related to the gastrointestinal tract.
A clinical study found gastrointestinal reactions to be more frequent and more severe in rheumatoid arthritis patients taking daily doses of 1500 mg naproxen compared to those taking 750 mg naproxen.
In controlled clinical trials with about 80 pediatric patients and in well-monitored, open-label studies with about 400 pediatric patients with polyarticular juvenile idiopathic arthritis treated with naproxen, the incidence of rash and prolonged bleeding times were greater, the incidence of gastrointestinal and central nervous system reactions were about the same, and the incidence of other reactions were lower in pediatric patients than in adults.
In patients taking naproxen in clinical trials, the most frequently reported adverse experiences in approximately 1% to 10% of patients were:
Gastrointestinal (GI) Experiences, including: heartburn*, abdominal pain*, nausea*, constipation*, diarrhea, dyspepsia, stomatitis
Central Nervous System: headache*, dizziness*, drowsiness*, lightheadedness, vertigo
Dermatologic: pruritus (itching)*, skin eruptions*, ecchymoses*, sweating, purpura
Special Senses: tinnitus*, visual disturbances, hearing disturbances
Cardiovascular: edema*, palpitations
General: dyspnea*, thirst
*Incidence of reported reaction between 3% and 9%. Those reactions occurring in less than 3% of the patients are unmarked.
In patients taking NSAIDs, the following adverse experiences have also been reported in approximately 1% to 10% of patients.
Gastrointestinal (GI) Experiences, including: flatulence, gross bleeding/perforation, GI ulcers (gastric/duodenal), vomiting
General: abnormal renal function, anemia, elevated liver enzymes, increased bleeding time, rashes
The following are additional adverse experiences reported in < 1% of patients taking naproxen during clinical trials.
Gastrointestinal: pancreatitis, vomiting
Hepatobiliary: jaundice
Hemic and Lymphatic: melena, thrombocytopenia, agranulocytosis
Nervous System: inability to concentrate
Dermatologic: skin rashes
Postmarketing ExperienceThe following adverse reactions have been identified during post approval use of naproxen. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following are additional adverse experiences reported in < 1% of patients taking naproxen during clinical trials and through postmarketing reports. Those adverse reactions observed through postmarketing reports are italicized.
Body as a Whole: anaphylactoid reactions, angioneurotic edema, menstrual disorders, pyrexia (chills and fever)
Cardiovascular: congestive heart failure, vasculitis, hypertension, pulmonary edema
Gastrointestinal: inflammation, bleeding (sometimes fatal, particularly in the elderly), ulceration, perforation and obstruction of the upper or lower gastrointestinal tract. Esophagitis, stomatitis, hematemesis, colitis, exacerbation of inflammatory bowel disease (ulcerative colitis, Crohn's disease).
Hepatobiliary: abnormal liver function tests, hepatitis (some cases have been fatal)
Hemic and Lymphatic: eosinophilia, leucopenia, granulocytopenia, hemolytic anemia, aplastic anemia
Metabolic and Nutritional: hyperglycemia, hypoglycemia
Nervous System: depression, dream abnormalities, insomnia, malaise, myalgia, muscle weakness, aseptic meningitis, cognitive dysfunction, convulsions
Respiratory: eosinophilic pneumonitis, asthma
Dermatologic: alopecia, urticaria, toxic epidermal necrolysis, erythema multiforme, erythema nodosum, fixed drug eruption, lichen planus, pustular reaction, systemic lupus erythematoses, bullous reactions, including Stevens-Johnson syndrome, photosensitive dermatitis, photosensitivity reactions, including rare cases resembling porphyria cutanea tarda (pseudoporphyria) or epidermolysis bullosa. If skin fragility, blistering or other symptoms suggestive of pseudoporphyria occur, treatment should be discontinued and the patient monitored.
Special Senses: hearing impairment, corneal opacity, papillitis, retrobulbar optic neuritis, papilledema
Urogenital: glomerular nephritis, hematuria, hyperkalemia, interstitial nephritis, nephrotic syndrome, renal disease, renal failure, renal papillary necrosis, raised serum creatinine
Reproduction (female): infertility
In patients taking NSAIDs, the following adverse experiences have also been reported in < 1% of patients.
Body as a Whole: fever, infection, sepsis, anaphylactic reactions, appetite changes, death
Cardiovascular: hypertension, tachycardia, syncope, arrhythmia, hypotension, myocardial infarction
Gastrointestinal: dry mouth, esophagitis, gastric/peptic ulcers, gastritis, glossitis, eructation
Hepatobiliary: hepatitis, liver failure
Hemic and Lymphatic: rectal bleeding, lymphadenopathy, pancytopenia
Metabolic and Nutritional: weight changes
Nervous System: anxiety, asthenia, confusion, nervousness, paresthesia, somnolence, tremors, convulsions, coma, hallucinations
Respiratory: asthma, respiratory depression, pneumonia
Dermatologic: exfoliative dermatitis
Special Senses: blurred vision, conjunctivitis
Urogenital: cystitis, dysuria, oliguria/polyuria, proteinuria
Therapeutic indications
NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS are indicated for:
the relief of the signs and symptoms of:
- rheumatoid arthritis
- osteoarthritis
- ankylosing spondylitis
- Polyarticular Juvenile Idiopathic Arthritis
NAPROSYN Tablets and ANAPROX DS are also indicated for:
the relief of signs and symptoms of:
- tendonitis
- bursitis
- acute gout
the management of:
- pain
- primary dysmenorrhea
Pharmacokinetic properties
Naproxen and naproxen sodium are rapidly and completely absorbed from the gastrointestinal tract with an in vivo bioavailability of 95%. The different dosage forms of NAPROSYN are bioequivalent in terms of extent of absorption (AUC) and peak concentration (Cmax); however, the products do differ in their pattern of absorption. These differences between naproxen products are related to both the chemical form of naproxen used and its formulation. Even with the observed differences in pattern of absorption, the elimination half-life of naproxen is unchanged across products ranging from 12 to 17 hours. Steady-state levels of naproxen are reached in 4 to 5 days, and the degree of naproxen accumulation is consistent with this half-life. This suggests that the differences in pattern of release play only a negligible role in the attainment of steady-state plasma levels.
AbsorptionNAPROSYN Tablets/ANAPROX DS: After administration of NAPROSYN Tablets, peak plasma levels are attained in 2 to 4 hours. After oral administration of ANAPROX DS, peak plasma levels are attained in 1 to 2 hours. The difference in rates between the two products is due to the increased aqueous solubility of the sodium salt of naproxen used in ANAPROX DS.
EC-NAPROSYN: EC-NAPROSYN is designed with a pH-sensitive coating to provide a barrier to disintegration in the acidic environment of the stomach and to lose integrity in the more neutral environment of the small intestine. The enteric polymer coating selected for EC-NAPROSYN dissolves above pH 6. When EC-NAPROSYN was given to fasted subjects, peak plasma levels were attained about 4 to 6 hours following the first dose (range: 2 to 12 hours). An in vivo study in man using radiolabeled ECNAPROSYN tablets demonstrated that EC-NAPROSYN dissolves primarily in the small intestine rather than in the stomach, so the absorption of the drug is delayed until the stomach is emptied.
When EC-NAPROSYN and NAPROSYN Tablets were given to fasted subjects (n=24) in a crossover study following 1 week of dosing, differences in time to peak plasma levels (Tmax) were observed, but there were no differences in total absorption as measured by Cmax and AUC:
| EC-NAPROSYN* 500 mg bid | NAPROSYN* 500 mg bid | |
| Cmax (μg/mL) | 94.9 (18%) | 97.4 (13%) |
| Tmax (hours) | 4 (39%) | 1.9 (61%) |
| AUC0-12 hr (μg•hr/mL) | 845 (20%) | 767 (15%) |
| *Mean value (coefficient of variation) |
Antacid Effects
When EC-NAPROSYN was given as a single dose with antacid (54 mEq buffering capacity), the peak plasma levels of naproxen were unchanged, but the time to peak was reduced (mean Tmax fasted 5.6 hours, mean Tmax with antacid 5 hours), although not significantly.
Food Effects
When EC-NAPROSYN was given as a single dose with food, peak plasma levels in most subjects were achieved in about 12 hours (range: 4 to 24 hours). Residence time in the small intestine until disintegration was independent of food intake. The presence of food prolonged the time the tablets remained in the stomach, time to first detectable serum naproxen levels, and time to maximal naproxen levels (Tmax), but did not affect peak naproxen levels (Cmax).
DistributionNaproxen has a volume of distribution of 0.16 L/kg. At therapeutic levels naproxen is greater than 99% albumin-bound. At doses of naproxen greater than 500 mg/day there is less than proportional increase in plasma levels due to an increase in clearance caused by saturation of plasma protein binding at higher doses (average trough Css 36.5, 49.2 and 56.4 mg/L with 500, 1000 and 1500 mg daily doses of naproxen, respectively). The naproxen anion has been found in the milk of lactating women at a concentration equivalent to approximately 1% of maximum naproxen concentration in plasma.
EliminationMetabolism
Naproxen is extensively metabolized in the liver to 6-0-desmethyl naproxen, and both parent and metabolites do not induce metabolizing enzymes. Both naproxen and 6-0-desmethyl naproxen are further metabolized to their respective acylglucuronide conjugated metabolites.
Excretion
The clearance of naproxen is 0.13 mL/min/kg. Approximately 95% of the naproxen from any dose is excreted in the urine, primarily as naproxen ( < 1%), 6-0-desmethyl naproxen ( < 1%) or their conjugates (66% to 92%). The plasma half-life of the naproxen anion in humans ranges from 12 to 17 hours. The corresponding half-lives of both naproxen's metabolites and conjugates are shorter than 12 hours, and their rates of excretion have been found to coincide closely with the rate of naproxen clearance from the plasma. Small amounts, 3% or less of the administered dose, are excreted in the feces. In patients with renal failure metabolites may accumulate.
Date of revision of the text
Aug 2016Name of the medicinal product
Naprosyn, Anaprox, Anaprox DSFertility, pregnancy and lactation
Risk SummaryUse of NSAIDs, including NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS, during the third trimester of pregnancy increases the risk of premature closure of the fetal ductus arteriosus. Avoid use of NSAIDs, including NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS, in pregnant women starting at 30 weeks of gestation (third trimester).
There are no adequate and well-controlled studies of NAPROSYN Tablets, EC-NAPROSYN or ANAPROX DS in pregnant women. Data from observational studies regarding potential embryofetal risks of NSAID use in women in the first or second trimesters of pregnancy are inconclusive. In the general U.S. population, all clinically recognized pregnancies, regardless of drug exposure, have a background rate of 2-4% for major malformations, and 15-20% for pregnancy loss. In animal reproduction studies in rats, rabbits, and mice no evidence of teratogenicity or fetal harm when naproxen was administered during the period of organogenesis at doses 0.13, 0.26, and 0.6 times the maximum recommended human daily dose of 1500 mg/day, respectively. Based on animal data, prostaglandins have been shown to have an important role in endometrial vascular permeability, blastocyst implantation, and decidualization. In animal studies, administration of prostaglandin synthesis inhibitors such as naproxen, resulted in increased pre-and post-implantation loss.
Clinical ConsiderationsLabor or Delivery
There are no studies on the effects of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS during labor or delivery. In animal studies, NSAIDS, including naproxen, inhibit prostaglandin synthesis, cause delayed parturition, and increase the incidence of stillbirth.
DataHuman Data
There is some evidence to suggest that when inhibitors of prostaglandin synthesis are used to delay preterm labor, there is an increased risk of neonatal complications such as necrotizing enterocolitis, patent ductus arteriosus, and intracranial hemorrhage. Naproxen treatment given in late pregnancy to delay parturition has been associated with persistent pulmonary hypertension, renal dysfunction, and abnormal prostaglandin E levels in preterm infants. Because of the known effects of nonsteroidal anti-inflammatory drugs on the fetal cardiovascular system (closure of ductus arteriosus), use during pregnancy (particularly starting at 30-weeks of gestation, or third trimester) should be avoided.
Animal data
Reproduction studies have been performed in rats at 20 mg/kg/day (0.13 times the maximum recommended human daily dose of 1500 mg/day based on body surface area comparison), rabbits at 20 mg/kg/day (0.26 times the maximum recommended human daily dose, based on body surface area comparison), and mice at 170 mg/kg/day (0.6 times the maximum recommended human daily dose based on body surface area comparison) with no evidence of impaired fertility or harm to the fetus due to the drug. Based on animal data, prostaglandins have been shown to have an important role in endometrial vascular permeability, blastocyst implantation, and decidualization. In animal studies, administration of prostaglandin synthesis inhibitors such as naproxen, resulted in increased pre-and post-implantation loss.
Qualitative and quantitative composition
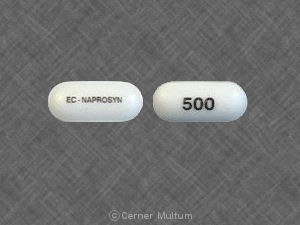
Dosage Forms And StrengthsNAPROSYN® (naproxen) tablets: 500 mg: yellow, capsule-shaped, engraved with NPR LE 500 on one side and scored on the other.
EC-NAPROSYN® (naproxen) delayed-release tablets: 375 mg: white, oval biconvex coated tablets imprinted with NPR EC 375 on one side.
EC-NAPROSYN® (naproxen) delayed-release tablets: 500 mg: white, oblong coated tablets imprinted with NPR EC 500 on one side.
ANAPROX® DS (naproxen sodium) tablets: 550 mg: dark blue, oblong-shaped, engraved with NPS 550 on one side and scored on both sides.
Storage And HandlingNAPROSYN (naproxen) tablets 500 mg: yellow, capsule-shaped tablets, engraved with NPR LE 500 on one side and scored on the other. Packaged in light-resistant bottles of 100. Supplied as:
NDC 69437-316-01 100's (bottle)
Store at 15°C to 30°C (59°F to 86°F) in well-closed containers; dispense in light-resistant containers.
EC-NAPROSYN (naproxen) delayed-release tablets 375 mg: white, oval biconvex coated tablets imprinted with NPR EC 375 on one side. Packaged in light-resistant bottles of 100. Supplied as:
NDC 69437-415-01 100's (bottle)
500 mg: white, oblong coated tablets imprinted with NPR EC 500 on one side. Packaged in light-resistant bottles of 100. Supplied as:
NDC 69437-416-01 100's (bottle)
Store at 15°C to 30°C (59°F to 86°F) in well-closed containers; dispense in light-resistant containers.
ANAPROX DS (naproxen sodium) Tablets 550 mg: dark blue, oblong-shaped tablets, engraved with NPS 550 on one side and scored on both sides. Packaged in bottles of 100. Supplied as:
NDC 69437-203-01 100's (bottle) Store at 15° to 30°C (59° to 86°F) in well-closed containers.
Manufactured for:Atnahs Pharma, Miles Gray Road, Basildon, Essex, SS14 3FR, United Kingdom Distributed by: Canton Laboratories, LLC, Alpharetta, GA 30004-5945, United States For more information, call 1-844-302-5227. Issued or Revised: Aug 2016
Special warnings and precautions for use
WARNINGSIncluded as part of the PRECAUTIONS section.
PRECAUTIONS Cardiovascular Thrombotic EventsClinical trials of several COX-2 selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, including myocardial infarction (MI) and stroke, which can be fatal. Based on available data, it is unclear that the risk for CV thrombotic events is similar for all NSAIDs. The relative increase in serious CV thrombotic events over baseline conferred by NSAID use appears to be similar in those with and without known CV disease or risk factors for CV disease. However, patients with known CV disease or risk factors had a higher absolute incidence of excess serious CV thrombotic events, due to their increased baseline rate. Some observational studies found that this increased risk of serious CV thrombotic events began as early as the first weeks of treatment. The increase in CV thrombotic risk has been observed most consistently at higher doses.
To minimize the potential risk for an adverse CV event in NSAID-treated patients, use the lowest effective dose for the shortest duration possible. Physicians and patients should remain alert for the development of such events, throughout the entire treatment course, even in the absence of previous CV symptoms. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID, such as naproxen, increases the risk of serious gastrointestinal (GI) events.
Status Post Coronary Artery Bypass Graft (CABG) SurgeryTwo large, controlled clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10-14 days following CABG surgery found an increased incidence of myocardial infarction and stroke. NSAIDs are contraindicated in the setting of CABG.
Post-MI PatientsObservational studies conducted in the Danish National Registry have demonstrated that patients treated with NSAIDs in the post-MI period were at increased risk of reinfarction, CV-related death, and all-cause mortality beginning in the first week of treatment. In this same cohort, the incidence of death in the first year post-MI was 20 per 100 person years in NSAID-treated patients compared to 12 per 100 person years in non-NSAID exposed patients. Although the absolute rate of death declined somewhat after the first year post-MI, the increased relative risk of death in NSAID users persisted over at least the next four years of follow-up.
Avoid the use of NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS in patients with a recent MI unless the benefits are expected to outweigh the risk of recurrent CV thrombotic events. If NAPROSYN Tablets, EC-NAPROSYN and ANAPROX DS are used in patients with a recent MI, monitor patients for signs of cardiac ischemia.
Gastrointestinal Bleeding, Ulceration, And PerforationNSAIDs, including naproxen, cause serious gastrointestinal (GI) adverse events including inflammation, bleeding, ulceration, and perforation of the esophagus, stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with NSAIDs.
Only one in five patients who develop a serious upper GI adverse event on NSAID therapy is symptomatic. Upper GI ulcers, gross bleeding, or perforation caused by NSAIDs occurred in approximately 1% of patients treated for 3-6 months, and in about 2%-4% of patients treated for one year. However, even short-term NSAID therapy is not without risk.
Risk Factors For GI Bleeding, Ulceration, And PerforationPatients with a prior history of peptic ulcer disease and/or GI bleeding who used NSAIDs had a greater than 10-fold increased risk for developing a GI bleed compared to patients without these risk factors. Other factors that increase the risk of GI bleeding in patients treated with NSAIDs include longer duration of NSAID therapy; concomitant use of oral corticosteroids, aspirin, anticoagulants, or selective serotonin reuptake inhibitors (SSRIs); smoking; use of alcohol; older age; and poor general health status. Most postmarketing reports of fatal GI events occurred in elderly or debilitated patients. Additionally, patients with advanced liver disease and/or coagulopathy are at increased risk for GI bleeding.
Strategies To Minimize The GI Risks In NSAID-treated Patients- Use the lowest effective dosage for the shortest possible duration.
- Avoid administration of more than one NSAID at a time.
- Avoid use in patients at higher risk unless benefits are expected to outweigh the increased risk of bleeding. For such patients, as well as those with active GI bleeding, consider alternate therapies other than NSAIDs.
- Remain alert for signs and symptoms of GI ulceration and bleeding during NSAID therapy.
- If a serious GI adverse event is suspected, promptly initiate evaluation and treatment, and discontinue NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS until a serious GI adverse event is ruled out.
- In the setting of concomitant use of low-dose aspirin for cardiac prophylaxis, monitor patients more closely for evidence of GI bleeding .
Elevations of ALT or AST (three or more times the upper limit of normal [ULN]) have been reported in approximately 1% of NSAID-treated patients in clinical trials. In addition, rare, sometimes fatal, cases of severe hepatic injury, including fulminant hepatitis, liver necrosis, and hepatic failure have been reported.
Elevations of ALT or AST (less than three times ULN) may occur in up to 15% of patients treated with NSAIDs including naproxen.
Inform patients of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, diarrhea, pruritus, jaundice, right upper quadrant tenderness, and “flu-like” symptoms). If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash, etc.), discontinue NAPROSYN Tablets, ECNAPROSYN, or ANAPROX DS immediately, and perform a clinical evaluation of the patient.
HypertensionNSAIDs, including NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS, can lead to new onset of hypertension or worsening of pre-existing hypertension, either of which may contribute to the increased incidence of CV events. Patients taking angiotensin converting enzyme (ACE) inhibitors, thiazide diuretics, or loop diuretics may have impaired response to these therapies when taking NSAIDs.
Monitor blood pressure (BP) during the initiation of NSAID treatment and throughout the course of therapy.
Heart Failure And EdemaThe Coxib and traditional NSAID Trialists' Collaboration meta-analysis of randomized controlled trials demonstrated an approximately two-fold increase in hospitalizations for heart failure in COX-2 selective-treated patients and nonselective NSAID-treated patients compared to placebo-treated patients. In a Danish National Registry study of patients with heart failure, NSAID use increased the risk of MI, hospitalization for heart failure, and death.
Additionally, fluid retention and edema have been observed in some patients treated with NSAIDs. Use of naproxen may blunt the CV effects of several therapeutic agents used to treat these medical conditions (e.g., diuretics, ACE inhibitors, or angiotensin receptor blockers [ARBs]).
Avoid the use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS in patients with severe heart failure unless the benefits are expected to outweigh the risk of worsening heart failure. If NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS is used in patients with severe heart failure, monitor patients for signs of worsening heart failure.
Since each ANAPROX DS tablet contains 50 mg of sodium (about 2 mEq per each 500 mg of naproxen), this should be considered in patients whose overall intake of sodium must be severely restricted.
Renal Toxicity And Hyperkalemia Renal ToxicityLong-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury.
Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of an NSAID may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, dehydration, hypovolemia, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors or ARBs, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
No information is available from controlled clinical studies regarding the use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS in patients with advanced renal disease. The renal effects of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS may hasten the progression of renal dysfunction in patients with preexisting renal disease.
Correct volume status in dehydrated or hypovolemic patients prior to initiating NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS. Monitor renal function in patients with renal or hepatic impairment, heart failure, dehydration, or hypovolemia during use of NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS. Avoid the use of NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS in patients with advanced renal disease unless the benefits are expected to outweigh the risk of worsening renal function. If NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS is used in patients with advanced renal disease, monitor patients for signs of worsening renal function.
HyperkalemiaIncreases in serum potassium concentration, including hyperkalemia, have been reported with use of NSAIDs, even in some patients without renal impairment. In patients with normal renal function, these effects have been attributed to a hyporeninemic-hypoaldosteronism state.
Anaphylactic ReactionsNaproxen has been associated with anaphylactic reactions in patients with and without known hypersensitivity to naproxen and in patients with aspirin-sensitive asthma.
Seek emergency help if an anaphylactic reaction occurs.
Exacerbation Of Asthma Related To Aspirin SensitivityA subpopulation of patients with asthma may have aspirin-sensitive asthma which may include chronic rhinosinusitis complicated by nasal polyps; severe, potentially fatal bronchospasm; and/or intolerance to aspirin and other NSAIDs.
Because cross-reactivity between aspirin and other NSAIDs has been reported in such aspirin-sensitive patients, NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS are contraindicated in patients with this form of aspirin sensitivity. When NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS is used in patients with preexisting asthma (without known aspirin sensitivity), monitor patients for changes in the signs and symptoms of asthma.
Serious Skin ReactionsNSAIDs, including naproxen, can cause serious skin adverse reactions such as exfoliative dermatitis, Stevens-Johnson Syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. These serious events may occur without warning. Inform patients about the signs and symptoms of serious skin reactions, and to discontinue the use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS at the first appearance of skin rash or any other sign of hypersensitivity. NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS are contraindicated in patients with previous serious skin reactions to NSAIDs.
Premature Closure Of Fetal Ductus ArteriosusNaproxen may cause premature closure of the fetal ductus arteriosus. Avoid use of NSAIDs, including NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS, in pregnant women starting at 30 weeks of gestation (third trimester).
Hematologic ToxicityAnemia has occurred in NSAID-treated patients. This may be due to occult or gross blood loss, fluid retention, or an incompletely described effect on erythropoiesis. If a patient treated with NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS has any signs or symptoms of anemia, monitor hemoglobin or hematocrit.
NSAIDs, including NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS, may increase the risk of bleeding events. Co-morbid conditions such as coagulation disorders or concomitant use of warfarin and other anticoagulants, antiplatelet agents (e.g., aspirin), serotonin reuptake inhibitors (SSRIs), and serotonin norepinephrine reuptake inhibitors (SNRIs) may increase this risk. Monitor these patients for signs of bleeding.
Masking Of Inflammation And FeverThe pharmacological activity of NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS in reducing inflammation, and possibly fever, may diminish the utility of diagnostic signs in detecting infections.
Long-Term Use And Laboratory MonitoringBecause serious GI bleeding, hepatotoxicity, and renal injury can occur without warning symptoms or signs, consider monitoring patients on long-term NSAID treatment with a CBC and a chemistry profile periodically.
Patients with initial hemoglobin values of 10g or less who are to receive long-term therapy should have hemoglobin values determined periodically.
Because of adverse eye findings in animal studies with drugs of this class, it is recommended that ophthalmic studies be carried out if any change or disturbance in vision occurs.
Patient Counseling InformationAdvise the patient to read the FDA-approved patient labeling (Medication Guide) that accompanies each prescription dispensed. Inform patients, families, or their caregivers of the following information before initiating therapy with NAPROSYN Tablets, EC-NAPROSYN or ANAPROX DS and periodically during the course of ongoing therapy.
Cardiovascular Thrombotic EventsAdvise patients to be alert for the symptoms of cardiovascular thrombotic events, including chest pain, shortness of breath, weakness, or slurring of speech, and to report any of these symptoms to their health care provider immediately.
Gastrointestinal Bleeding, Ulceration, And PerforationAdvise patients to report symptoms of ulcerations and bleeding, including epigastric pain, dyspepsia, melena, and hematemesis to their health care provider. In the setting of concomitant use of low-dose aspirin for cardiac prophylaxis, inform patients of the increased risk for and the signs and symptoms of GI bleeding.
HepatotoxicityInform patients of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, diarrhea, jaundice, right upper quadrant tenderness, and “flu-like” symptoms). If these occur, instruct patients to stop NAPROSYN Tablets, EC-NAPROSYN or ANAPROX DS and seek immediate medical therapy.
Heart Failure And EdemaAdvise patients to be alert for the symptoms of congestive heart failure including shortness of breath, unexplained weight gain, or edema and to contact their healthcare provider if such symptoms occur .
Anaphylactic ReactionsInform patients of the signs of an anaphylactic reaction (e.g., difficulty breathing, swelling of the face or throat). Instruct patients to seek immediate emergency help if these occur.
Serious Skin ReactionsAdvise patients to stop NAPROSYN Tablets, EC-NAPROSYN or ANAPROX DS immediately if they develop any type of rash and to contact their healthcare provider as soon as possible.
Female FertilityAdvise females of reproductive potential who desire pregnancy that NSAIDs, including NAPROSYN Tablets, ECNAPROSYN, and ANAPROX DS, may be associated with a reversible delay in ovulation (see Use in Specific Populations.)
Fetal ToxicityInform pregnant women to avoid use of NAPROSYN Tablets, EC-NAPROSYN or ANAPROX DS and other NSAIDs starting at 30 weeks gestation because of the risk of the premature closing of the fetal ductus arteriosus.
Avoid Concomitant Use Of NSAIDsInform patients that the concomitant use of NAPROSYN Tablets, EC-NAPROSYN and ANAPROX DS with other NSAIDs or salicylates (e.g., diflunisal, salsalate) is not recommended due to the increased risk of gastrointestinal toxicity, and little or no increase in efficacy. Alert patients that NSAIDs may be present in “over the counter” medications for treatment of colds, fever, or insomnia.
Use Of NSAIDS And Low-Dose AspirinInform patients not to use low-dose aspirin concomitantly with NAPROSYN Tablets, EC-NAPROSYN and ANAPROX DS until they talk to their healthcare provider.
Nonclinical Toxicology Carcinogenesis, Mutagenesis, Impairment Of Fertility CarcinogenesisA 2-year study was performed in rats to evaluate the carcinogenic potential of naproxen at rat doses of 8, 16, and 24 mg/kg/day (0.05, 0.1, and 0.16 times the maximum recommended human daily dose [MRHD] of 1500 mg/day based on a body surface area comparison). No evidence of tumorigenicity was found.
MutagenesisNaproxen tested positive in the in vivo sister chromatid exchange assay for but was not mutagenic in the in vitro bacterial reverse mutation assay (Ames test).
Impairment Of FertilityMale rats were treated with 2, 5, 10, and 20 mg/kg naproxen by oral gavage for 60 days prior to mating and female rats were treated with the same doses for 14 days prior to mating and for the first 7 days of pregnancy. There were no adverse effects on fertility noted (up to 0.13 times the MRDH based on body surface area).
Use In Specific Populations Pregnancy Risk SummaryUse of NSAIDs, including NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS, during the third trimester of pregnancy increases the risk of premature closure of the fetal ductus arteriosus. Avoid use of NSAIDs, including NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS, in pregnant women starting at 30 weeks of gestation (third trimester).
There are no adequate and well-controlled studies of NAPROSYN Tablets, EC-NAPROSYN or ANAPROX DS in pregnant women. Data from observational studies regarding potential embryofetal risks of NSAID use in women in the first or second trimesters of pregnancy are inconclusive. In the general U.S. population, all clinically recognized pregnancies, regardless of drug exposure, have a background rate of 2-4% for major malformations, and 15-20% for pregnancy loss. In animal reproduction studies in rats, rabbits, and mice no evidence of teratogenicity or fetal harm when naproxen was administered during the period of organogenesis at doses 0.13, 0.26, and 0.6 times the maximum recommended human daily dose of 1500 mg/day, respectively. Based on animal data, prostaglandins have been shown to have an important role in endometrial vascular permeability, blastocyst implantation, and decidualization. In animal studies, administration of prostaglandin synthesis inhibitors such as naproxen, resulted in increased pre-and post-implantation loss.
Clinical ConsiderationsLabor or Delivery
There are no studies on the effects of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS during labor or delivery. In animal studies, NSAIDS, including naproxen, inhibit prostaglandin synthesis, cause delayed parturition, and increase the incidence of stillbirth.
DataHuman Data
There is some evidence to suggest that when inhibitors of prostaglandin synthesis are used to delay preterm labor, there is an increased risk of neonatal complications such as necrotizing enterocolitis, patent ductus arteriosus, and intracranial hemorrhage. Naproxen treatment given in late pregnancy to delay parturition has been associated with persistent pulmonary hypertension, renal dysfunction, and abnormal prostaglandin E levels in preterm infants. Because of the known effects of nonsteroidal anti-inflammatory drugs on the fetal cardiovascular system (closure of ductus arteriosus), use during pregnancy (particularly starting at 30-weeks of gestation, or third trimester) should be avoided.
Animal data
Reproduction studies have been performed in rats at 20 mg/kg/day (0.13 times the maximum recommended human daily dose of 1500 mg/day based on body surface area comparison), rabbits at 20 mg/kg/day (0.26 times the maximum recommended human daily dose, based on body surface area comparison), and mice at 170 mg/kg/day (0.6 times the maximum recommended human daily dose based on body surface area comparison) with no evidence of impaired fertility or harm to the fetus due to the drug. Based on animal data, prostaglandins have been shown to have an important role in endometrial vascular permeability, blastocyst implantation, and decidualization. In animal studies, administration of prostaglandin synthesis inhibitors such as naproxen, resulted in increased pre-and post-implantation loss.
Lactation Risk SummaryThe naproxen anion has been found in the milk of lactating women at a concentration equivalent to approximately 1% of maximum naproxen concentration in plasma. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and any potential adverse effects on the breastfed infant from the NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS or from the underlying maternal condition.
Females And Males Of Reproductive Potential InfertilityFemales
Based on the mechanism of action, the use of prostaglandin-mediated NSAIDs, including NAPROSYN Tablets, ECNAPROSYN, and ANAPROX DS, may delay or prevent rupture of ovarian follicles, which has been associated with reversible infertility in some women. Published animal studies have shown that administration of prostaglandin synthesis inhibitors has the potential to disrupt prostaglandin-mediated follicular rupture required for ovulation. Small studies in women treated with NSAIDs have also shown a reversible delay in ovulation. Consider withdrawal of NSAIDs, including NAPROSYN Tablets, EC-NAPROSYN and ANAPROX DS, in women who have difficulties conceiving or who are undergoing investigation of infertility.
Pediatric UseSafety and effectiveness in pediatric patients below the age of 2 years have not been established. Pediatric dosing recommendations for polyarticular juvenile idiopathic arthritis are based on well-controlled studies. There are no adequate effectiveness or dose-response data for other pediatric conditions, but the experience in polyarticular juvenile idiopathic arthritis and other use experience have established that single doses of 2.5 to 5 mg/kg as naproxen suspension, , with total daily dose not exceeding 15 mg/kg/day, are well tolerated in pediatric patients over 2 years of age.
Geriatric UseThe hepatic and renal tolerability of long-term naproxen administration was studied in two double-blind clinical trials involving 586 patients. Of the patients studied, 98 patients were age 65 and older and 10 of the 98 patients were age 75 and older. NAPROXEN was administered at doses of 375 mg twice daily or 750 mg twice daily for up to 6 months. Transient abnormalities of laboratory tests assessing hepatic and renal function were noted in some patients, although there were no differences noted in the occurrence of abnormal values among different age groups.
Elderly patients, compared to younger patients, are at greater risk for NSAID-associated serious cardiovascular, gastrointestinal, and/or renal adverse reactions. If the anticipated benefit for the elderly patient outweighs these potential risks, start dosing at the low end of the dosing range, and monitor patients for adverse effects.
Studies indicate that although total plasma concentration of naproxen is unchanged, the unbound plasma fraction of naproxen is increased in the elderly. The clinical significance of this finding is unclear, although it is possible that the increase in free naproxen concentration could be associated with an increase in the rate of adverse events per a given dosage in some elderly patients. Caution is advised when high doses are required and some adjustment of dosage may be required in elderly patients. As with other drugs used in the elderly, it is prudent to use the lowest effective dose.
Experience indicates that geriatric patients may be particularly sensitive to certain adverse effects of nonsteroidal anti-inflammatory drugs. Elderly or debilitated patients seem to tolerate peptic ulceration or bleeding less well when these events do occur. Most spontaneous reports of fatal GI events are in the geriatric population.
Naproxen is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. Geriatric patients may be at a greater risk for the development of a form of renal toxicity precipitated by reduced prostaglandin formation during administration of nonsteroidal anti-inflammatory drugs.
Hepatic ImpairmentCaution is advised when high doses are required and some adjustment of dosage may be required in these patients. It is prudent to use the lowest effective dose.
Renal ImpairmentNaproxen-containing products are not recommended for use in patients with moderate to severe and severe renal impairment (creatinine clearance < 30 mL/min).
Dosage (Posology) and method of administration
General Dosing InstructionsCarefully consider the potential benefits and risks of NAPROSYN Tablets, EC-NAPROSYN and ANAPROX DS and other treatment options before deciding to use NAPROSYN Tablets, EC-NAPROSYN and ANAPROX DS. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals.
After observing the response to initial therapy with NAPROSYN Tablets, EC-NAPROSYN or ANAPROX DS, the dose and frequency should be adjusted to suit an individual patient's needs.
To maintain the integrity of the enteric coating, the EC-NAPROSYN tablet should not be broken, crushed or chewed during ingestion.
Naproxen-containing products such as NAPROSYN, EC-NAPROSYN and ANAPROX DS, and other naproxen products should not be used concomitantly since they all circulate in the plasma as the naproxen anion.
Rheumatoid Arthritis, Osteoarthritis And Ankylosing SpondylitisThe recommended dosages of NAPROSYN Tablets, ANAPROX DS, and EC-NAPROSYN are shown in Table 1.
Table 1: Recommended dosages for NAPROSYN Tablets,
ANAPROX DS, and EC-NAPROSYN
| NAPROSYN | 250 mg (one half tablet) 500 mg | twice daily |
| ANAPROX DS | 275 mg (one half tablet) 550 mg (naproxen 500 mg with 50 mg sodium) | twice daily |
| EC-NAPROSYN | 375 mg | twice daily |
| or 500 mg | twice daily |
During long-term administration, the dose of naproxen may be adjusted up or down depending on the clinical response of the patient. A lower daily dose may suffice for long-term administration. The morning and evening doses do not have to be equal in size and the administration of the drug more frequently than twice daily is not necessary.
The morning and evening doses do not have to be equal in size and administration of the drug more frequently than twice daily does not generally make a difference in response.
In patients who tolerate lower doses well, the dose may be increased to naproxen 1500 mg/day for limited periods of up to 6 months when a higher level of anti-inflammatory/analgesic activity is required. When treating such patients with naproxen 1500 mg/day, the physician should observe sufficient increased clinical benefits to offset the potential increased risk.
Polyarticular Juvenile Idiopathic ArthritisNaproxen solid-oral dosage forms may not allow for the flexible dose titration needed in pediatric patients with polyarticular juvenile idiopathic arthritis. A liquid formulation may be more appropriate for weight-based dosing and due to the need for dose flexibility in children.
In pediatric patients, doses of 5 mg/kg/day produced plasma levels of naproxen similar to those seen in adults taking 500 mg of naproxen. The recommended total daily dose of naproxen is approximately 10 mg/kg given in 2 divided doses. Dosing with NAPROSYN Tablets is not appropriate for children weighing less than 50 kilograms.
Management Of Pain, Primary Dysmenorrhea, And Acute Tendonitis And BursitisThe recommended starting dose of ANAPROX DS (naproxen sodium) tablets is 550 mg followed by 550 mg every 12 hours or 275 mg (one half of a 550 mg tablet) every 6 to 8 hours as required. The initial total daily dose should not exceed 1375 mg (two and one-half tablets) of naproxen sodium. Thereafter, the total daily dose should not exceed 1100 mg of naproxen sodium. Because the sodium salt of naproxen is more rapidly absorbed, ANAPROX DS is recommended for the management of acute painful conditions when prompt onset of pain relief is desired. NAPROSYN Tablets may also be used. The recommended starting dose of NAPROSYN Tablets is 500 mg followed by 250 mg (one half of a 500 mg NAPROSYN tablet) every 6-8 hours as required.. The total daily dose should not exceed 1250 mg of naproxen.
EC-NAPROSYN is not recommended for initial treatment of acute pain because absorption of naproxen is delayed compared to other naproxen-containing products.
Acute GoutThe recommended starting dose is 750 mg (one and one-half tablets) of NAPROSYN Tablets followed by 250 mg (one-half tablet) every 8 hours until the attack has subsided. ANAPROX DS may also be used at a starting dose of 825 mg (one and one-half tablets) followed by 275 mg (one-half tablet) every 8 hours. EC-NAPROSYN is not recommended because of the delay in absorption.
Non-Interchangeability With Other Formulations Of NaproxenDifferent dose strengths and formulations (e.g., tablets, suspension) of naproxen are not interchangeable. This difference should be taken into consideration when changing strengths or formulations.
Interaction with other medicinal products and other forms of interaction
SIDE EFFECTSThe following adverse reactions are discussed in greater detail in other sections of the labeling:
- Cardiovascular Thrombotic Events
- GI Bleeding, Ulceration, and Perforation
- Hepatotoxicity
- Hypertension
- Heart Failure and Edema
- Renal Toxicity and Hyperkalemia
- Anaphylactic Reactions
- Serious Skin Reactions
- Hematologic Toxicity
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse reactions reported in controlled clinical trials in 960 patients treated for rheumatoid arthritis or osteoarthritis are listed below. In general, reactions in patients treated chronically were reported 2 to 10 times more frequently than they were in short-term studies in the 962 patients treated for mild to moderate pain or for dysmenorrhea. The most frequent complaints reported related to the gastrointestinal tract.
A clinical study found gastrointestinal reactions to be more frequent and more severe in rheumatoid arthritis patients taking daily doses of 1500 mg naproxen compared to those taking 750 mg naproxen.
In controlled clinical trials with about 80 pediatric patients and in well-monitored, open-label studies with about 400 pediatric patients with polyarticular juvenile idiopathic arthritis treated with naproxen, the incidence of rash and prolonged bleeding times were greater, the incidence of gastrointestinal and central nervous system reactions were about the same, and the incidence of other reactions were lower in pediatric patients than in adults.
In patients taking naproxen in clinical trials, the most frequently reported adverse experiences in approximately 1% to 10% of patients were:
Gastrointestinal (GI) Experiences, including: heartburn*, abdominal pain*, nausea*, constipation*, diarrhea, dyspepsia, stomatitis
Central Nervous System: headache*, dizziness*, drowsiness*, lightheadedness, vertigo
Dermatologic: pruritus (itching)*, skin eruptions*, ecchymoses*, sweating, purpura
Special Senses: tinnitus*, visual disturbances, hearing disturbances
Cardiovascular: edema*, palpitations
General: dyspnea*, thirst
*Incidence of reported reaction between 3% and 9%. Those reactions occurring in less than 3% of the patients are unmarked.
In patients taking NSAIDs, the following adverse experiences have also been reported in approximately 1% to 10% of patients.
Gastrointestinal (GI) Experiences, including: flatulence, gross bleeding/perforation, GI ulcers (gastric/duodenal), vomiting
General: abnormal renal function, anemia, elevated liver enzymes, increased bleeding time, rashes
The following are additional adverse experiences reported in < 1% of patients taking naproxen during clinical trials.
Gastrointestinal: pancreatitis, vomiting
Hepatobiliary: jaundice
Hemic and Lymphatic: melena, thrombocytopenia, agranulocytosis
Nervous System: inability to concentrate
Dermatologic: skin rashes
Postmarketing ExperienceThe following adverse reactions have been identified during post approval use of naproxen. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following are additional adverse experiences reported in < 1% of patients taking naproxen during clinical trials and through postmarketing reports. Those adverse reactions observed through postmarketing reports are italicized.
Body as a Whole: anaphylactoid reactions, angioneurotic edema, menstrual disorders, pyrexia (chills and fever)
Cardiovascular: congestive heart failure, vasculitis, hypertension, pulmonary edema
Gastrointestinal: inflammation, bleeding (sometimes fatal, particularly in the elderly), ulceration, perforation and obstruction of the upper or lower gastrointestinal tract. Esophagitis, stomatitis, hematemesis, colitis, exacerbation of inflammatory bowel disease (ulcerative colitis, Crohn's disease).
Hepatobiliary: abnormal liver function tests, hepatitis (some cases have been fatal)
Hemic and Lymphatic: eosinophilia, leucopenia, granulocytopenia, hemolytic anemia, aplastic anemia
Metabolic and Nutritional: hyperglycemia, hypoglycemia
Nervous System: depression, dream abnormalities, insomnia, malaise, myalgia, muscle weakness, aseptic meningitis, cognitive dysfunction, convulsions
Respiratory: eosinophilic pneumonitis, asthma
Dermatologic: alopecia, urticaria, toxic epidermal necrolysis, erythema multiforme, erythema nodosum, fixed drug eruption, lichen planus, pustular reaction, systemic lupus erythematoses, bullous reactions, including Stevens-Johnson syndrome, photosensitive dermatitis, photosensitivity reactions, including rare cases resembling porphyria cutanea tarda (pseudoporphyria) or epidermolysis bullosa. If skin fragility, blistering or other symptoms suggestive of pseudoporphyria occur, treatment should be discontinued and the patient monitored.
Special Senses: hearing impairment, corneal opacity, papillitis, retrobulbar optic neuritis, papilledema
Urogenital: glomerular nephritis, hematuria, hyperkalemia, interstitial nephritis, nephrotic syndrome, renal disease, renal failure, renal papillary necrosis, raised serum creatinine
Reproduction (female): infertility
In patients taking NSAIDs, the following adverse experiences have also been reported in < 1% of patients.
Body as a Whole: fever, infection, sepsis, anaphylactic reactions, appetite changes, death
Cardiovascular: hypertension, tachycardia, syncope, arrhythmia, hypotension, myocardial infarction
Gastrointestinal: dry mouth, esophagitis, gastric/peptic ulcers, gastritis, glossitis, eructation
Hepatobiliary: hepatitis, liver failure
Hemic and Lymphatic: rectal bleeding, lymphadenopathy, pancytopenia
Metabolic and Nutritional: weight changes
Nervous System: anxiety, asthenia, confusion, nervousness, paresthesia, somnolence, tremors, convulsions, coma, hallucinations
Respiratory: asthma, respiratory depression, pneumonia
Dermatologic: exfoliative dermatitis
Special Senses: blurred vision, conjunctivitis
Urogenital: cystitis, dysuria, oliguria/polyuria, proteinuria
DRUG INTERACTIONSSee Table 1 for clinically significant drug interactions with naproxen.
Table 1: Clinically Significant Drug Interactions with
naproxen
| Drugs That Interfere with Hemostasis | |
| Clinical Impact: |
|
| Intervention: | Monitor patients with concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS with anticoagulants (e.g., warfarin), antiplatelet agents (e.g., aspirin), selective serotonin reuptake inhibitors (SSRIs), and serotonin norepinephrine reuptake inhibitors (SNRIs) for signs of bleeding . |
| Aspirin | |
| Clinical Impact: | Controlled clinical studies showed that the concomitant use of NSAIDs and analgesic doses of aspirin does not produce any greater therapeutic effect than the use of NSAIDs alone. In a clinical study, the concomitant use of an NSAID and aspirin was associated with a significantly increased incidence of GI adverse reactions as compared to use of the NSAID alone. |
| Intervention: | Concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and analgesic doses of aspirin is not generally recommended because of the increased risk of bleeding. NAPROSYN Tablets, EC-NAPROSYN, and ANAPROX DS are not substitutes for low dose aspirin for cardiovascular protection. |
| ACE Inhibitors, Angiotensin Receptor Blockers, and Beta-Blockers | |
| Clinical Impact: |
|
| Intervention: |
|
| Diuretics | |
| Clinical Impact: | Clinical studies, as well as post-marketing observations, showed that NSAIDs reduced the natriuretic effect of loop diuretics (e.g., furosemide) and thiazide diuretics in some patients. This effect has been attributed to the NSAID inhibition of renal prostaglandin synthesis. |
| Intervention: | During concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS with diuretics, observe patients for signs of worsening renal function, in addition to assuring diuretic efficacy including antihypertensive effects. |
| Digoxin | |
| Clinical Impact: | The concomitant use of naproxen with digoxin has been reported to increase the serum concentration and prolong the half-life of digoxin |
| Intervention: | During concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and digoxin, monitor serum digoxin levels. |
| Lithium | |
| Clinical Impact: | NSAIDs have produced elevations in plasma lithium levels and reductions in renal lithium clearance. The mean minimum lithium concentration increased 15%, and the renal clearance decreased by approximately 20%. This effect has been attributed to NSAID inhibition of renal prostaglandin synthesis. |
| Intervention: | During concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and lithium, monitor patients for signs of lithium toxicity. |
| Methotrexate | |
| Clinical Impact: | Concomitant use of NSAIDs and methotrexate may increase the risk for methotrexate toxicity (e.g., neutropenia, thrombocytopenia, renal dysfunction). |
| Intervention: | During concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and methotrexate, monitor patients for methotrexate toxicity. |
| Cyclosporine | |
| Clinical Impact: | Concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and cyclosporine may increase cyclosporine’s nephrotoxicity. |
| Intervention: | During concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and cyclosporine, monitor patients for signs of worsening renal function. |
| NSAIDs and Salicylates | |
| Clinical Impact: | Concomitant use of naproxen with other NSAIDs or salicylates (e.g., diflunisal, salsalate) increases the risk of GI toxicity, with little or no increase in efficacy. |
| Intervention: | The concomitant use of naproxen with other NSAIDs or salicylates is not recommended. |
| Pemetrexed | |
| Clinical Impact: | Concomitant use of NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and pemetrexed may increase the risk of pemetrexed-associated myelosuppression, renal, and GI toxicity (see the pemetrexed prescribing information). |
| Intervention: | During concomitant use of NAPROSYN Tablets, EC-NAPROSYN or ANAPROX DS and pemetrexed, in patients with renal impairment whose creatinine clearance ranges from 45 to 79 mL/min, monitor for myelosuppression, renal and GI toxicity. NSAIDs with short elimination half-lives (e.g., diclofenac, indomethacin) should be avoided for a period of two days before, the day of, and two days following administration of pemetrexed. In the absence of data regarding potential interaction between pemetrexed and NSAIDs with longer half-lives (e.g., meloxicam, nabumetone), patients taking these NSAIDs should interrupt dosing for at least five days before, the day of, and two days following pemetrexed administration. |
| Antacids and Sucralfate | |
| Clinical Impact: | Concomitant administration of some antacids (magnesium oxide or aluminum hydroxide) and sucralfate can delay the absorption of naproxen. |
| Intervention: | Concomitant administration of antacids such as magnesium oxide or aluminum hydroxide, and sucralfate with NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS is not recommended. |
| Cholestyramine | |
| Clinical Impact: | Concomitant administration of cholestyramine can delay the absorption of naproxen. |
| Intervention: | Concomitant administration of cholestyramine with NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS is not recommended. |
| Probenecid | |
| Clinical Impact: | Probenecid given concurrently increases naproxen anion plasma levels and extends its plasma half-life significantly. |
| Intervention: | Patients simultaneously receiving NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and probenecid should be observed for adjustment of dose if required. |
| Other albumin-bound drugs | |
| Clinical Impact: | Naproxen is highly bound to plasma albumin; it thus has a theoretical potential for interaction with other albumin-bound drugs such as coumarin-type anticoagulants, sulphonylureas, hydantoins, other NSAIDs, and aspirin. |
| Intervention: | Patients simultaneously receiving NAPROSYN Tablets, EC-NAPROSYN, or ANAPROX DS and a hydantoin, sulphonamide or sulphonylurea should be observed for adjustment of dose if required. |
| Bleeding times | |
| Clinical Impact: | Naproxen may decrease platelet aggregation and prolong bleeding time. |
| Intervention: | This effect should be kept in mind when bleeding times are determined. |
| Porter-Silber test | |
| Clinical Impact: | The administration of naproxen may result in increased urinary values for 17-ketogenic steroids because of an interaction between the drug and/or its metabolites with m-di-nitrobenzene used in this assay. |
| Intervention: | Although 17-hydroxy-corticosteroid measurements (Porter-Silber test) do not appear to be artifactually altered, it is suggested that therapy with naproxen be temporarily discontinued 72 hours before adrenal function tests are performed if the Porter-Silber test is to be used. |
| Urinary assays of 5-hydroxy indoleacetic acid (5HIAA) | |
| Clinical Impact: | Naproxen may interfere with some urinary assays of 5-hydroxy indoleacetic acid (5HIAA). |
| Intervention: | This effect should be kept in mind when urinary 5-hydroxy indoleacetic acid is determined. |