Gleevec
Drugs with the same active ingredients
Overdose
Experience with doses greater than 800 mg is limited. Isolated cases of Gleevec overdose have been reported. In the event of overdosage, observe the patient and give appropriate supportive treatment.
Adult Overdose1,200 to 1,600 mg (duration varying between 1 to 10 days): Nausea, vomiting, diarrhea, rash erythema, edema, swelling, fatigue, muscle spasms, thrombocytopenia, pancytopenia, abdominal pain, headache, decreased appetite.
1,800 to 3,200 mg (as high as 3,200 mg daily for 6 days): Weakness, myalgia, increased CPK, increased bilirubin, gastrointestinal pain.
6,400 mg (single dose): One case in the literature reported one patient who experienced nausea, vomiting, abdominal pain, pyrexia, facial swelling, neutrophil count decreased, increase transaminases.
8 to 10 g (single dose): Vomiting and gastrointestinal pain have been reported.
A patient with myeloid blast crisis experienced Grade 1 elevations of serum creatinine, Grade 2 ascites and elevated liver transaminase levels, and Grade 3 elevations of bilirubin after inadvertently taking 1,200 mg of Gleevec daily for 6 days. Therapy was temporarily interrupted and complete reversal of all abnormalities occurred within 1 week. Treatment was resumed at a dose of 400 mg daily without recurrence of adverse reactions. Another patient developed severe muscle cramps after taking 1,600 mg of Gleevec daily for 6 days. Complete resolution of muscle cramps occurred following interruption of therapy and treatment was subsequently resumed. Another patient that was prescribed 400 mg daily, took 800 mg of Gleevec on Day 1 and 1,200 mg on Day 2. Therapy was interrupted, no adverse reactions occurred and the patient resumed therapy.
Pediatric OverdoseOne 3-year-old male exposed to a single dose of 400 mg experienced vomiting, diarrhea and anorexia and another 3-year-old male exposed to a single dose of 980 mg experienced decreased white blood cell count and diarrhea.
Contraindications
None.
Undesirable effects
The following serious adverse reactions are described elsewhere in the labeling:
- Fluid Retention and Edema
- Hematologic Toxicity
- Congestive Heart Failure and Left Ventricular Dysfunction
- Hepatotoxicity
- Hemorrhage
- Gastrointestinal Disorders
- Hypereosinophilic Cardiac Toxicity
- Dermatologic Toxicities
- Hypothyroidism
- Growth Retardation in Children and Adolescents
- Tumor Lysis Syndrome
- Impairments Related to Driving and Using Machinery
- Renal Toxicity
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Chronic Myeloid LeukemiaThe majority of Gleevec-treated patients experienced adverse reactions at some time. Gleevec was discontinued due to drug-related adverse reactions in 2.4% of patients receiving Gleevec in the randomized trial of newly diagnosed patients with Ph+ CML in chronic phase comparing Gleevec versus IFN+Ara-C, and in 12.5% of patients receiving Gleevec in the randomized trial of newly diagnosed patients with Ph+ CML in chronic phase comparing Gleevec and nilotinib. Gleevec was discontinued due to drug-related adverse reactions in 4% of patients in chronic phase after failure of interferon-alpha therapy, in 4% of patients in accelerated phase and in 5% of patients in blast crisis.
The most frequently reported drug-related adverse reactions were edema, nausea and vomiting, muscle cramps, musculoskeletal pain, diarrhea and rash (Table 2 and Table 3 for newly diagnosed CML, Table 4 for other CML patients). Edema was most frequently periorbital or in lower limbs and was managed with diuretics, other supportive measures, or by reducing the dose of Gleevec. The frequency of severe superficial edema was 1.5%-6%.
A variety of adverse reactions represent local or general fluid retention including pleural effusion, ascites, pulmonary edema and rapid weight gain with or without superficial edema. These reactions appear to be dose related, were more common in the blast crisis and accelerated phase studies (where the dose was 600 mg/day), and are more common in the elderly. These reactions were usually managed by interrupting Gleevec treatment and using diuretics or other appropriate supportive care measures. These reactions may be serious or life threatening.
Adverse reactions, regardless of relationship to study drug, that were reported in at least 10% of the Gleevec treated patients are shown in Tables 2, 3, and 4.
Table 2: Adverse Reactions Regardless of Relationship
to Study Drug Reported in Newly Diagnosed CML Clinical Trial in the Gleevec
versus IFN+Ara-C Study (greater than or equal to 10% of Gleevec Treated
Patients)(1)
| Preferred Term | All Grades | CTC Grades 3/4 | ||
| Gleevec N=551 (%) |
IFN+Ara-C N=533 (%) |
Gleevec N=551 (%) |
IFN+Ara-C N=533 (%) |
|
| Fluid Retention | 61.7 | 11.1 | 2.5 | 0.9 |
| - Superficial Edema | 59.9 | 9.6 | 1.5 | 0.4 |
| - Other Fluid Retention Reactions2 | 6.9 | 1.9 | 1.3 | 0.6 |
| Nausea | 49.5 | 61.5 | 1.3 | 5.1 |
| Muscle Cramps | 49.2 | 11.8 | 2.2 | 0.2 |
| Musculoskeletal Pain | 47.0 | 44.8 | 5.4 | 8.6 |
| Diarrhea | 45.4 | 43.3 | 3.3 | 3.2 |
| Rash and Related Terms | 40.1 | 26.1 | 2.9 | 2.4 |
| Fatigue | 38.8 | 67.0 | 1.8 | 25.1 |
| Headache | 37.0 | 43.3 | 0.5 | 3.8 |
| Joint Pain | 31.4 | 38.1 | 2.5 | 7.7 |
| Abdominal Pain | 36.5 | 25.9 | 4.2 | 3.9 |
| Nasopharyngitis | 30.5 | 8.8 | 0 | 0.4 |
| Hemorrhage | 28.9 | 21.2 | 1.8 | 1.7 |
| - GI Hemorrhage | 1.6 | 1.1 | 0.5 | 0.2 |
| - CNS Hemorrhage | 0.2 | 0.4 | 0 | 0.4 |
| Myalgia | 24.1 | 38.8 | 1.5 | 8.3 |
| Vomiting | 22.5 | 27.8 | 2.0 | 3.4 |
| Dyspepsia | 18.9 | 8.3 | 0 | 0.8 |
| Cough | 20.0 | 23.1 | 0.2 | 0.6 |
| Pharyngolaryngeal Pain | 18.1 | 11.4 | 0.2 | 0 |
| Upper Respiratory Tract Infection | 21.2 | 8.4 | 0.2 | 0.4 |
| Dizziness | 19.4 | 24.4 | 0.9 | 3.8 |
| Pyrexia | 17.8 | 42.6 | 0.9 | 3.0 |
| Weight Increased | 15.6 | 2.6 | 2.0 | 0.4 |
| Insomnia | 14.7 | 18.6 | 0 | 2.3 |
| Depression | 14.9 | 35.8 | 0.5 | 13.1 |
| Influenza | 13.8 | 6.2 | 0.2 | 0.2 |
| Bone Pain | 11.3 | 15.6 | 1.6 | 3.4 |
| Constipation | 11.4 | 14.4 | 0.7 | 0.2 |
| Sinusitis | 11.4 | 6.0 | 0.2 | 0.2 |
| (1)All adverse reactions occurring in greater than or
equal to10% of Gleevec treated patients are listed regardless of suspected
relationship to treatment. (2)Other fluid retention reactions include pleural effusion, ascites, pulmonary edema, pericardial effusion, anasarca, edema aggravated, and fluid retention not otherwise specified. |
Table 3: Most Frequently Reported Non-hematologic
Adverse Reactions (Regardless of Relationship to Study Drug) in Patients with
Newly Diagnosed Ph+ CML-CP in the Gleevec versus nilotinib Study (greater than
or equal to 10% in Gleevec 400 mg Once-Daily or nilotinib 300 mg Twice-Daily
Groups) 60-Month Analysisa
| Body System and Preferred Term | Patients with Newly Diagnosed Ph+ CML-CP | ||||
| Gleevec 400 mg once daily N=280 |
nilotinib 300 mg twice daily N=279 |
Gleevec 400 mg once daily N=280 |
nilotinib 300 mg twice daily N=279 |
||
| All Grades (%) | CTC Gradesb 3/4 (%) | ||||
| Skin and subcutaneous tissue disorders | Rash | 19 | 38 | 2 | < 1 |
| Pruritus | 7 | 21 | 0 | < 1 | |
| Alopecia | 7 | 13 | 0 | 0 | |
| Dry skin | 6 | 12 | 0 | 0 | |
| Gastrointestinal disorders | Nausea | 41 | 22 | 2 | 2 |
| Constipation | 8 | 20 | 0 | < 1 | |
| Diarrhea | 46 | 19 | 4 | 1 | |
| Vomiting | 27 | 15 | < 1 | < 1 | |
| Abdominal pain | 14 | 18 | < 1 | 1 | |
| upper Abdominal pain | 12 | 15 | 0 | 2 | |
| Dyspepsia | 12 | 10 | 0 | 0 | |
| Nervous system disorders | Headache | 23 | 32 | < 1 | 3 |
| Dizziness | 11 | 12 | < 1 | < 1 | |
| General disorders and administration site conditions | Fatigue | 20 | 23 | 1 | 1 |
| Pyrexia | 13 | 14 | 0 | <1 | |
| Asthenia | 12 | 14 | 0 | < 1 | |
| Peripheral edema | 20 | 9 | 0 | < 1 | |
| Face edema | 14 | < 1 | < 1 | 0 | |
| Musculoskeletal and connective tissue disorders | Myalgia | 19 | 19 | < 1 | < 1 |
| Arthralgia | 17 | 22 | < 1 | < 1 | |
| Muscle spasms | 34 | 12 | 1 | 0 | |
| Pain in extremity | 16 | 15 | < 1 | < 1 | |
| Back pain | 17 | 19 | 1 | 1 | |
| Respiratory, thoracic and mediastinal disorders | Cough | 13 | 17 | 0 | 0 |
| Oropharyngeal pain | 6 | 12 | 0 | 0 | |
| Dyspnea | 6 | 11 | < 1 | 2 | |
| Infections and infestations | Nasopharyngitis | 21 | 27 | 0 | 0 |
| Upper respiratory tract infection | 14 | 17 | 0 | < 1 | |
| Influenza | 9 | 13 | 0 | 0 | |
| Gastroenteritis | 10 | 7 | < 1 | 0 | |
| Eye disorders | Eyelid edema | 19 | 1 | < 1 | 0 |
| Periorbital edema | 15 | < 1 | 0 | 0 | |
| Psychiatric disorders | Insomnia | 9 | 11 | 0 | 0 |
| Vascular disorder | Hypertension | 4 | 10 | < 1 | 1 |
| aExcluding laboratory abnormalities bNCI Common Terminology Criteria for Adverse Events, Version 3.0 |
|||||
Table 4: Adverse Reactions Regardless of Relationship
to Study Drug Reported in Other CML Clinical Trials (greater than or equal to
10% of All Patients in any Trial)(1)
| Preferred Term | Myeloid Blast Crisis (n=260) % |
Accelerated Phase (n=235)% |
Chronic Phase, IFN Failure (n=532)% |
|||
| All Grades | Grade 3/4 | All Grades | Grade 3/4 | All Grades | Grade 3/4 | |
| Fluid Retention | 72 | 11 | 76 | 6 | 69 | 4 |
| -Superficial Edema | 66 | 6 | 74 | 3 | 67 | 2 |
| -Other Fluid Retention Reactions (2) | 22 | 6 | 15 | 4 | 7 | 2 |
| Nausea | 71 | 5 | 73 | 5 | 63 | 3 |
| Muscle Cramps | 28 | 1 | 47 | 0.4 | 62 | 2 |
| Vomiting | 54 | 4 | 58 | 3 | 36 | 2 |
| Diarrhea | 43 | 4 | 57 | 5 | 48 | 3 |
| Hemorrhage | 53 | 19 | 49 | 11 | 30 | 2 |
| - CNS Hemorrhage | 9 | 7 | 3 | 3 | 2 | 1 |
| - GI Hemorrhage | 8 | 4 | 6 | 5 | 2 | 0.4 |
| Musculoskeletal Pain | 42 | 9 | 49 | 9 | 38 | 2 |
| Fatigue | 30 | 4 | 46 | 4 | 48 | 1 |
| Skin Rash | 36 | 5 | 47 | 5 | 47 | 3 |
| Pyrexia | 41 | 7 | 41 | 8 | 21 | 2 |
| Arthralgia | 25 | 5 | 34 | 6 | 40 | 1 |
| Headache | 27 | 5 | 32 | 2 | 36 | 0.6 |
| Abdominal Pain | 30 | 6 | 33 | 4 | 32 | 1 |
| Weight Increased | 5 | 1 | 17 | 5 | 32 | 7 |
| Cough | 14 | 0.8 | 27 | 0.9 | 20 | 0 |
| Dyspepsia | 12 | 0 | 22 | 0 | 27 | 0 |
| Myalgia | 9 | 0 | 24 | 2 | 27 | 0.2 |
| Nasopharyngitis | 10 | 0 | 17 | 0 | 22 | 0.2 |
| Asthenia | 18 | 5 | 21 | 5 | 15 | 0.2 |
| Dyspnea | 15 | 4 | 21 | 7 | 12 | 0.9 |
| Upper Respiratory Tract Infection | 3 | 0 | 12 | 0.4 | 19 | 0 |
| Anorexia | 14 | 2 | 17 | 2 | 7 | 0 |
| Night Sweats | 13 | 0.8 | 17 | 1 | 14 | 0.2 |
| Constipation | 16 | 2 | 16 | 0.9 | 9 | 0.4 |
| Dizziness | 12 | 0.4 | 13 | 0 | 16 | 0.2 |
| Pharyngitis | 10 | 0 | 12 | 0 | 15 | 0 |
| Insomnia | 10 | 0 | 14 | 0 | 14 | 0.2 |
| Pruritus | 8 | 1 | 14 | 0.9 | 14 | 0.8 |
| Hypokalemia | 13 | 4 | 9 | 2 | 6 | 0.8 |
| Pneumonia | 13 | 7 | 10 | 7 | 4 | 1 |
| Anxiety | 8 | 0.8 | 12 | 0 | 8 | 0.4 |
| Liver Toxicity | 10 | 5 | 12 | 6 | 6 | 3 |
| Rigors | 10 | 0 | 12 | 0.4 | 10 | 0 |
| Chest Pain | 7 | 2 | 10 | 0.4 | 11 | 0.8 |
| Influenza | 0.8 | 0.4 | 6 | 0 | 11 | 0.2 |
| Sinusitis | 4 | 0.4 | 11 | 0.4 | 9 | 0.4 |
| (1)All adverse reactions occurring in greater than or
equal to10% of patients are listed regardless of suspected relationship to
treatment. (2)Other fluid retention reactions include pleural effusion, ascites, pulmonary edema, pericardial effusion, anasarca, edema aggravated, and fluid retention not otherwise specified. |
Cytopenias, and particularly neutropenia and thrombocytopenia, were a consistent finding in all studies, with a higher frequency at doses greater than or equal to 750 mg (Phase 1 study). The occurrence of cytopenias in CML patients was also dependent on the stage of the disease.
In patients with newly diagnosed CML, cytopenias were less frequent than in the other CML patients (see Tables 5, 6, and 7). The frequency of Grade 3 or 4 neutropenia and thrombocytopenia was between 2-and 3fold higher in blast crisis and accelerated phase compared to chronic phase (see Tables 4 and 5). The median duration of the neutropenic and thrombocytopenic episodes varied from 2 to 3 weeks, and from 2 to 4 weeks, respectively.
These reactions can usually be managed with either a reduction of the dose or an interruption of treatment with Gleevec, but may require permanent discontinuation of treatment.
Table 5: Laboratory Abnormalities in Newly Diagnosed
CML Clinical Trial (Gleevec versus IFN+Ara-C)
| CTC Grades | Gleevec N=551 % |
IFN+Ara-C N=533 % |
||
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | |
| Hematology Parameters* | ||||
| - Neutropenia* | 13.1 | 3.6 | 20.8 | 4.5 |
| - Thrombocytopenia* | 8.5 | 0.4 | 15.9 | 0.6 |
| - Anemia | 3.3 | 1.1 | 4.1 | 0.2 |
| Biochemistry Parameters | ||||
| - Elevated Creatinine | 0 | 0 | 0.4 | 0 |
| - Elevated Bilirubin | 0.9 | 0.2 | 0.2 | 0 |
| - Elevated Alkaline Phosphatase | 0.2 | 0 | 0.8 | 0 |
| - Elevated SGOT /SGPT | 4.7 | 0.5 | 7.1 | 0.4 |
| *p less than 0.001 (difference in Grade 3 plus 4 abnormalities between the two treatment groups) | ||||
Table 6: Percent Incidence
of Clinically Relevant Grade 3/4* Laboratory Abnormalities in the Newly
Diagnosed CML Clinical Trial (Gleevec versus nilotinib)
| Gleevec 400 mg once-daily N=280 (%) |
nilotinib 300 mg twice-daily N=279 (%) |
|
| Hematologic Parameters | ||
| Thrombocytopenia | 9 | 10 |
| Neutropenia | 22 | 12 |
| Anemia | 6 | 4 |
| Biochemistry Parameters | ||
| Elevated lipase | 4 | 9 |
| Hyperglycemia | < 1 | 7 |
| Hypophosphatemia | 10 | 8 |
| Elevated bilirubin (total) | < 1 | 4 |
| Elevated SGPT (ALT) | 3 | 4 |
| Hyperkalemia | 1 | 2 |
| Hyponatremia | < 1 | 1 |
| Hypokalemia | 2 | < 1 |
| Elevated SGOT (AST) | 1 | 1 |
| Decreased albumin | < 1 | 0 |
| Hypocalcemia | < 1 | < 1 |
| Elevated alkaline phosphatase | < 1 | 0 |
| Elevated creatinine | < 1 | 0 |
| *NCI Common Terminology Criteria for Adverse Events, version 3.0 | ||
Table 7: Laboratory Abnormalities in Other CML
Clinical Trials
| CTC Grades1 | Myeloid Blast Crisis (n=260) 600 mg n=223 400 mg n=37% |
Accelerated Phase (n=235) 600 mg n=158 400 mg n=77% |
Chronic Phase, IFN Failure (n=532) 400 mg% |
|||
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | Grade 3 | Grade 4 | |
| Hematology Parameters | ||||||
| - Neutropenia | 16 | 48 | 23 | 36 | 27 | 9 |
| - Thrombocytopenia | 30 | 33 | 31 | 13 | 21 | < 1 |
| - Anemia | 42 | 11 | 34 | 7 | 6 | 1 |
| Biochemistry Parameters | ||||||
| - Elevated Creatinine | 1.5 | 0 | 1.3 | 0 | 0.2 | 0 |
| - Elevated Bilirubin | 3.8 | 0 | 2.1 | 0 | 0.6 | 0 |
| - Elevated Alkaline Phosphatase | 4.6 | 0 | 5.5 | 0.4 | 0.2 | 0 |
| - Elevated SGOT (AST) | 1.9 | 0 | 3.0 | 0 | 2.3 | 0 |
| - Elevated SGPT (ALT) | 2.3 | 0.4 | 4.3 | 0 | 2.1 | 0 |
| 1CTC Grades: neutropenia (Grade 3 greater than or equal to 0.5-1.0 x 109/L, Grade 4 less than 0.5 x 109/L), thrombocytopenia (Grade 3 greater than or equal to 10-50 x 109/L, Grade 4 less than 10 x 109/L), anemia (hemoglobin greater than or equal to 65-80 g/L, Grade 4 less than 65 g/L), elevated creatinine (Grade 3 greater than 3-6 x upper limit normal range [ULN], Grade 4 greater than 6 x ULN), elevated bilirubin (Grade 3 greater than 3-10 x ULN, Grade 4 greater than 10 x ULN), elevated alkaline phosphatase (Grade 3 greater than 5-20 x ULN, Grade 4 greater than 20 x ULN), elevated SGOT or SGPT (Grade 3 greater than 5-20 x ULN, Grade 4 greater than 20 x ULN) | ||||||
Severe elevation of transaminases or bilirubin occurred in approximately 5% of CML patients (see Tables 6 and 7) and were usually managed with dose reduction or interruption (the median duration of these episodes was approximately 1 week). Treatment was discontinued permanently because of liver laboratory abnormalities in less than 1.0% of CML patients. One patient, who was taking acetaminophen regularly for fever, died of acute liver failure. In the Phase 2 GIST trial, Grade 3 or 4 SGPT (ALT) elevations were observed in 6.8% of patients and Grade 3 or 4 SGOT (AST) elevations were observed in 4.8% of patients. Bilirubin elevation was observed in 2.7% of patients.
Adverse Reactions In Pediatric Population Single Agent TherapyThe overall safety profile of pediatric patients treated with Gleevec in 93 children studied was similar to that found in studies with adult patients, except that musculoskeletal pain was less frequent (20.5%) and peripheral edema was not reported. Nausea and vomiting were the most commonly reported individual adverse reactions with an incidence similar to that seen in adult patients. Most patients experienced adverse reactions at some time during the study. The incidence of Grade 3/4 events across all types of adverse reactions was 75%; the events with the highest Grade 3/4 incidence in CML pediatric patients were mainly related to myelosuppression.
In Combination With Multi-Agent ChemotherapyPediatric and young adult patients with very high risk ALL, defined as those with an expected 5 year event-free survival (EFS) less than 45%, were enrolled after induction therapy on a multicenter, non-randomized cooperative group pilot protocol. The study population included patients with a median age of 10 years (1 to 21 years), 61% of whom were male, 75% were white, 7% were black and 6% were Asian/Pacific Islander. Patients with Ph+ ALL (n=92) were assigned to receive Gleevec and treated in 5 successive cohorts. Gleevec exposure was systematically increased in successive cohorts by earlier introduction and more prolonged duration.
The safety of Gleevec given in combination with intensive chemotherapy was evaluated by comparing the incidence of grade 3 and 4 adverse events, neutropenia (less than 750/mcL) and thrombocytopenia (less than 75,000/mcL) in the 92 patients with Ph+ ALL compared to 65 patients with Ph-ALL enrolled on the trial who did not receive Gleevec. The safety was also evaluated comparing the incidence of adverse events in cycles of therapy administered with or without Gleevec. The protocol included up to 18 cycles of therapy. Patients were exposed to a cumulative total of 1425 cycles of therapy, 778 with Gleevec and 647 without Gleevec. The adverse events that were reported with a 5% or greater incidence in patients with Ph+ ALL compared to Ph-ALL or with a 1% or greater incidence in cycles of therapy that included Gleevec are presented in Table 8.
Table 8: Adverse Reactions Reported More Frequently in
Patients Treated with Study Drug (greater than 5%) or in Cycles with Study Drug
(greater than 1%)
| Adverse Event | Per Patient Incidence Ph+ALL With Gleevec N=92 n (%) |
Per Patient Incidence Ph- ALL No Gleevec N=65 n (%) |
Per Patient Per Cycle Incidence With Gleevec* N=778 n (%) |
Per Patient Per Cycle Incidence No Gleevec** N=647 n (%) |
| Grade 3 and 4 Adverse Events Nausea and/or Vomiting | 15 (16) | 6 (9) | 28 (4) | 8 (1) |
| Hypokalemia | 31 (34) | 16 (25) | 72 (9) | 32(5) |
| Pneumonitis | 7 (8) | 1 (1) | 7(1) | 1(< 1) |
| Pleural effusion | 6 (7) | 0 | 6 (1) | 0 |
| Abdominal Pain | 8 (9) | 2 (3) | 9 (1) | 3(< 1) |
| Anorexia | 10 (11) | 3 (5) | 19 (2) | 4 (1) |
| Hemorrhage | 11 (12) | 4 (6) | 17 (2) | 8 (1) |
| Hypoxia | 8 (9) | 2 (3) | 12 (2) | 2 (< 1) |
| Myalgia | 5 (5) | 0 | 4 (1) | 1 (< 1) |
| Stomatitis | 15 (16) | 8 (12) | 22 (3) | 14 (2) |
| Diarrhea | 8 (9) | 3 (5) |
Therapeutic indications
Newly Diagnosed Philadelphia Positive Chronic Myeloid Leukemia (Ph+ CML)Newly diagnosed adult and pediatric patients with Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase.
Ph+ CML In Blast Crisis (BC), Accelerated Phase (AP) Or Chronic Phase (CP) After Interferon-alpha (IFN) TherapyPatients with Philadelphia chromosome positive chronic myeloid leukemia in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy.
Adult Patients With Ph+ Acute Lymphoblastic Leukemia (ALL)Adult patients with relapsed or refractory Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL).
Pediatric Patients With Ph+ Acute Lymphoblastic Leukemia (ALL)Pediatric patients with newly diagnosed Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL) in combination with chemotherapy.
Myelodysplastic/Myeloproliferative Diseases (MDS/MPD)Adult patients with myelodysplastic/myeloproliferative diseases associated with PDGFR (platelet-derived growth factor receptor) gene re-arrangements as determined with an FDA-approved test.
Aggressive Systemic Mastocytosis (ASM)Adult patients with aggressive systemic mastocytosis without the D816V c-Kit mutation as determined with an FDA-approved test or with c-Kit mutational status unknown.
Hypereosinophilic Syndrome (HES) And/Or Chronic Eosinophilic Leukemia (CEL)Adult patients with hypereosinophilic syndrome and/or chronic eosinophilic leukemia who have the FIP1L1-PDGFRα fusion kinase (mutational analysis or FISH demonstration of CHIC2 allele deletion) and for patients with HES and/or CEL who are FIP1L1-PDGFRα fusion kinase negative or unknown.
Dermatofibrosarcoma Protuberans (DFSP)Adult patients with unresectable, recurrent and/or metastatic dermatofibrosarcoma protuberans.
Kit+ Gastrointestinal Stromal Tumors (GIST)Patients with Kit (CD117) positive unresectable and/or metastatic malignant gastrointestinal stromal tumors.
Adjuvant Treatment of GISTAdjuvant treatment of adult patients following complete gross resection of Kit (CD117) positive GIST.
Pharmacokinetic properties
The pharmacokinetics of Gleevec have been evaluated in studies in healthy subjects and in population pharmacokinetic studies in over 900 patients. The pharmacokinetics of Gleevec are similar in CML and GIST patients.
Absorption And DistributionImatinib is well absorbed after oral administration with Cmax achieved within 2-4 hours post-dose. Mean absolute bioavailability is 98%. Mean imatinib AUC increases proportionally with increasing doses ranging from 25 mg to 1,000 mg. There is no significant change in the pharmacokinetics of imatinib on repeated dosing, and accumulation is 1.5-to 2.5-fold at steady state when Gleevec is dosed once-daily. At clinically relevant concentrations of imatinib, binding to plasma proteins in in vitro experiments is approximately 95%, mostly to albumin and α1-acid glycoprotein.
EliminationMetabolism
CYP3A4 is the major enzyme responsible for metabolism of imatinib. Other cytochrome P450 enzymes, such as CYP1A2, CYP2D6, CYP2C9, and CYP2C19, play a minor role in its metabolism. The main circulating active metabolite in humans is the N-demethylated piperazine derivative, formed predominantly by CYP3A4. It shows in vitro potency similar to the parent imatinib. The plasma AUC for this metabolite is about 15% of the AUC for imatinib. The plasma protein binding of N-demethylated metabolite CGP74588 is similar to that of the parent compound. Human liver microsome studies demonstrated that Gleevec is a potent competitive inhibitor of CYP2C9, CYP2D6, and CYP3A4/5 with Ki values of 27, 7.5, and 8 μM, respectively.
Excretion
Imatinib elimination is predominately in the feces, mostly as metabolites. Based on the recovery of compound(s) after an oral 14C-labeled dose of imatinib, approximately 81% of the dose was eliminated within 7 days, in feces (68% of dose) and urine (13% of dose). Unchanged imatinib accounted for 25% of the dose (5% urine, 20% feces), the remainder being metabolites.
Following oral administration in healthy volunteers, the elimination half-lives of imatinib and its major active metabolite, the N-demethyl derivative (CGP74588), are approximately 18 and 40 hours, respectively.
Typically, clearance of imatinib in a 50-year-old patient weighing 50 kg is expected to be 8 L/h, while for a 50year-old patient weighing 100 kg the clearance will increase to 14 L/h. The inter-patient variability of 40% in clearance does not warrant initial dose adjustment based on body weight and/or age but indicates the need for close monitoring for treatment-related toxicity.
Date of revision of the text
Sep 2017Name of the medicinal product
GleevecFertility, pregnancy and lactation
Risk SummaryGleevec can cause fetal harm when administered to a pregnant woman based on human and animal data. There are no clinical studies regarding use of Gleevec in pregnant women. There have been postmarket reports of spontaneous abortions and congenital anomalies from women who have been exposed to Gleevec during pregnancy. Reproductive studies in rats have demonstrated that imatinib mesylate induced teratogenicity and increased incidence of congenital abnormalities following prenatal exposure to imatinib mesylate at doses equal to the highest recommended human dose of 800 mg/day based on body surface area. Advise women to avoid pregnancy when taking Gleevec. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is not known; however, in the U.S. general population, the estimated background risk of major birth defects of clinically recognized pregnancies is 2-4% and of miscarriage is 15%-20%.
DataAnimal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of imatinib mesylate up to 100 mg/kg/day and 60 mg/kg/day, respectively, during the period of organogenesis.
In rats, imatinib mesylate was teratogenic at 100 mg/kg/day (approximately equal to the maximum human dose of 800 mg/day based on body surface area), the number of fetuses with encephalocoele and exencephaly was higher than historical control values and these findings were associated with missing or underdeveloped cranial bones. Lower mean fetal body weights were associated with retarded skeletal ossifications.
In rabbits, at doses 1.5 times higher than the maximum human dose of 800 mg/day based on body surface area, no effects on the reproductive parameters with respect to implantation sites, number of live fetuses, sex ratio or fetal weight were observed. The examinations of the fetuses did not reveal any drug related morphological changes.
In a pre-and postnatal development study in rats, pregnant rats received oral doses of imatinib mesylate during gestation (organogenesis) and lactation up to 45 mg/kg/day. Five animals developed a red vaginal discharge in the 45 mg/kg/day group on Days 14 or 15 of gestation, the significance of which is unknown since all females produced viable litters and none had increased post-implantation loss. Other maternal effects noted only at the dose of 45 mg/kg/day (approximately one-half the maximum human dose of 800 mg/day based on body surface area) included an increased numbers of stillborn pups and pups dying between postpartum Days 0 and 4. In the F1 offspring at this same dose level, mean body weights were reduced from birth until terminal sacrifice and the number of litters achieving criterion for preputial separation was slightly decreased. There were no other significant effects in developmental parameters or behavioral testing. F1 fertility was not affected but reproductive effects were noted at 45 mg/kg/day including an increased number of resorptions and a decreased number of viable fetuses. The NOEL for both maternal animals and the F1 generation was 15 mg/kg/day.
Qualitative and quantitative composition
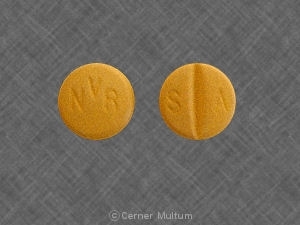
Dosage Forms And Strengths 100 mg film coated tabletsVery dark yellow to brownish orange, film-coated tablets, round, biconvex with bevelled edges, debossed with “NVR” on one side, and “SA” with score on the other side
400 mg film coated tabletsVery dark yellow to brownish orange, film-coated tablets, ovaloid, biconvex with bevelled edges, debossed with “400” on one side with score on the other side, and “SL” on each side of the score
400 mg film coated tabletsVery dark yellow to brownish orange, film-coated tablets, ovaloid, biconvex with bevelled edges, debossed with “gleevec” on one side and score on the other side.
Storage And HandlingEach film-coated tablet contains 100 mg or 400 mg of imatinib free base.
100 mg TabletsVery dark yellow to brownish orange, film-coated tablets, round, biconvex with bevelled edges, debossed with
“NVR” on one side, and “SA” with score on the other side.
Bottles of 90 tablets..........NDC 0078-0401-34
400 mg TabletsVery dark yellow to brownish orange, film-coated tablets, ovaloid, biconvex with bevelled edges, debossed with “400” on one side with score on the other side, and “SL” on each side of the score.
Bottles of 30 tablets..........NDC 0078-0438-15
400 mg TabletsVery dark yellow to brownish orange, film-coated tablets, ovaloid, biconvex with bevelled edges, debossed with “gleevec” on one side and score on the other side.
Unit Dose (blister pack of 30)..........NDC 0078-0649-30
Storage And HandlingStore at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F). Protect from moisture.
Dispense in a tight container, USP.
Do not crush Gleevec tablets. Avoid direct contact of crushed tablets with the skin or mucous membranes. If such contact occurs, wash thoroughly as outlined in the references. Avoid exposure to crushed tablets.
Distributed by: Novartis Pharmaceuticals Corporation East Hanover, New Jersey 07936. Revised: Sep 2017
Special warnings and precautions for use
WARNINGSIncluded as part of the PRECAUTIONS section.
PRECAUTIONS Fluid Retention And EdemaGleevec is often associated with edema and occasionally serious fluid retention. Weigh and monitor patients regularly for signs and symptoms of fluid retention. Investigate unexpected rapid weight gain carefully and provide appropriate treatment. The probability of edema was increased with higher Gleevec dose and age greater than 65 years in the CML studies. Severe superficial edema was reported in 1.5% of newly diagnosed CML patients taking Gleevec, and in 2%-6% of other adult CML patients taking Gleevec. In addition, other severe fluid retention (e.g., pleural effusion, pericardial effusion, pulmonary edema, and ascites) reactions were reported in 1.3% of newly diagnosed CML patients taking Gleevec, and in 2%-6% of other adult CML patients taking Gleevec. Severe fluid retention was reported in 9% to 13.1% of patients taking Gleevec for GIST. In a randomized trial in patients with newly diagnosed Ph+CML in chronic phase comparing Gleevec and nilotinib, severe (Grade 3 or 4) fluid retention occurred in 2.5% of patients receiving Gleevec and in 3.9% of patients receiving nilotinib 300 mg twice daily. Effusions (including pleural effusion, pericardial effusion, ascites) or pulmonary edema were observed in 2.1% (none were Grade 3 or 4) of patients in the Gleevec arm and 2.2% (0.7% Grade 3 or 4) of patients in the nilotinib 300 mg twice daily arm.
Hematologic ToxicityTreatment with Gleevec is associated with anemia, neutropenia, and thrombocytopenia. Perform complete blood counts weekly for the first month, biweekly for the second month, and periodically thereafter as clinically indicated (for example, every 2 to 3 months). In CML, the occurrence of these cytopenias is dependent on the stage of disease and is more frequent in patients with accelerated phase CML or blast crisis than in patients with chronic phase CML. In pediatric CML patients the most frequent toxicities observed were Grade 3 or 4 cytopenias including neutropenia, thrombocytopenia and anemia. These generally occur within the first several months of therapy.
Congestive Heart Failure And Left Ventricular DysfunctionCongestive heart failure and left ventricular dysfunction have been reported in patients taking Gleevec. Cardiac adverse reactions were more frequent in patients with advanced age or co-morbidities including previous medical history of cardiac disease. In an international randomized Phase 3 study in 1,106 patients with newly diagnosed Ph+ CML in chronic phase, severe cardiac failure and left ventricular dysfunction were observed in 0.7% of patients taking Gleevec compared to 0.9% of patients taking IFN + Ara-C. In another randomized trial with newly diagnosed Ph+ CML patients in chronic phase that compared Gleevec and nilotinib, cardiac failure was observed in 1.1% of patient in the Gleevec arm and 2.2% of patients in the nilotinib 300 mg twice daily arm and severe (Grade 3 or 4) cardiac failure occurred in 0.7% of patients in each group. Carefully monitor patients with cardiac disease or risk factors for cardiac or history of renal failure. Evaluate and treat any patient with signs or symptoms consistent with cardiac or renal failure.
HepatotoxicityHepatotoxicity, occasionally severe, may occur with Gleevec. Cases of fatal liver failure and severe liver injury requiring liver transplants have been reported with both short-term and long-term use of Gleevec. Monitor liver function (transaminases, bilirubin, and alkaline phosphatase) before initiation of treatment and monthly, or as clinically indicated. Manage laboratory abnormalities with Gleevec interruption and/or dose reduction.When Gleevec is combined with chemotherapy, liver toxicity in the form of transaminase elevation and hyperbilirubinemia has been observed. Additionally, there have been reports of acute liver failure. Monitoring of hepatic function is recommended.
HemorrhageIn a trial of Gleevec versus IFN+Ara-C in patients with the newly diagnosed CML, 1.8% of patients had Grade 3/4 hemorrhage. In the Phase 3 unresectable or metastatic GIST studies, 211 patients (12.9%) reported Grade 3/4 hemorrhage at any site. In the Phase 2 unresectable or metastatic GIST study, 7 patients (5%) had a total of 8 CTC Grade 3/4 hemorrhages; gastrointestinal (GI) (3 patients), intra-tumoral (3 patients) or both (1 patient). Gastrointestinal tumor sites may have been the source of GI hemorrhages. In a randomized trial in patients with newly diagnosed Ph+ CML in chronic phase comparing Gleevec and nilotinib, GI hemorrhage occurred in 1.4% of patients in the Gleevec arm, and in 2.9% of patients in the nilotinib 300 mg twice daily arm. None of these events were Grade 3 or 4 in the Gleevec arm; 0.7% were Grade 3 or 4 in the nilotinib 300 mg twice daily arm. In addition, gastric antral vascular ectasia has been reported in postmarketing experience.
Gastrointestinal DisordersGleevec is sometimes associated with GI irritation. Gleevec should be taken with food and a large glass of water to minimize this problem. There have been rare reports, including fatalities, of gastrointestinal perforation.
Hypereosinophilic Cardiac ToxicityIn patients with hypereosinophilic syndrome with occult infiltration of HES cells within the myocardium, cases of cardiogenic shock/left ventricular dysfunction have been associated with HES cell degranulation upon the initiation of Gleevec therapy. The condition was reported to be reversible with the administration of systemic steroids, circulatory support measures and temporarily withholding Gleevec.
Myelodysplastic/myeloproliferative disease and systemic mastocytosis may be associated with high eosinophil levels. Consider performing an echocardiogram and determining serum troponin in patients with HES/CEL, and in patients with MDS/MPD or ASM associated with high eosinophil levels. If either is abnormal, consider prophylactic use of systemic steroids (1–2 mg/kg) for one to two weeks concomitantly with Gleevec at the initiation of therapy.
Dermatologic ToxicitiesBullous dermatologic reactions, including erythema multiforme and Stevens-Johnson syndrome, have been reported with use of Gleevec. In some cases of bullous dermatologic reactions, including erythema multiforme and Stevens-Johnson syndrome reported during postmarketing surveillance, a recurrent dermatologic reaction was observed upon rechallenge. Several foreign postmarketing reports have described cases in which patients tolerated the reintroduction of Gleevec therapy after resolution or improvement of the bullous reaction. In these instances, Gleevec was resumed at a dose lower than that at which the reaction occurred and some patients also received concomitant treatment with corticosteroids or antihistamines.
HypothyroidismClinical cases of hypothyroidism have been reported in thyroidectomy patients undergoing levothyroxine replacement during treatment with Gleevec. Monitor TSH levels in such patients.
Embryo-fetal ToxicityGleevec can cause fetal harm when administered to a pregnant woman. Imatinib mesylate was teratogenic in rats when administered during organogenesis at doses approximately equal to the maximum human dose of 800 mg/day based on body surface area. Significant post-implantation loss was seen in female rats administered imatinib mesylate at doses approximately one-half the maximum human dose of 800 mg/day based on body surface area. Advise sexually active female patients of reproductive potential to use effective contraception (methods that result in less than 1% pregnancy rates) when using Gleevec and for 14 days after stopping Gleevec. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Growth Retardation In Children And AdolescentsGrowth retardation has been reported in children and pre-adolescents receiving Gleevec. The long term effects of prolonged treatment with Gleevec on growth in children are unknown. Therefore, monitor growth in children under Gleevec treatment.
Tumor Lysis SyndromeCases of Tumor Lysis Syndrome (TLS), including fatal cases, have been reported in patients with CML, GIST, ALL and eosinophilic leukemia receiving Gleevec. The patients at risk of TLS are those with tumors having a high proliferative rate or high tumor burden prior to treatment. Monitor these patients closely and take appropriate precautions. Due to possible occurrence of TLS, correct clinically significant dehydration and treat high uric acid levels prior to initiation of Gleevec.
Impairments Related To Driving And Using MachineryMotor vehicle accidents have been reported in patients receiving Gleevec. Advise patients that they may experience side effects such as dizziness, blurred vision or somnolence during treatment with Gleevec. Recommend caution when driving a car or operating machinery.
Renal ToxicityA decline in renal function may occur in patients receiving Gleevec. Median estimated glomerular filtration rate (eGFR) values in patients on Gleevec 400 mg daily for newly-diagnosed CML (four randomized trials) and malignant GIST (one single-arm trial) declined from a baseline value of 85 ml/min/1.73m² (N=1190) to 75 ml/min/1.73m² at 12 months (N=1082) and 69 ml/min/1.73m² at 60 months (N=549). Evaluate renal function prior to initiating Gleevec and monitor during therapy, with attention to risk factors for renal dysfunction such as pre-existing renal impairment, diabetes mellitus, hypertension, and congestive heart failure.
Nonclinical Toxicology Carcinogenesis, Mutagenesis, Impairment Of FertilityIn the 2-year rat carcinogenicity study administration of imatinib at 15, 30, and 60 mg/kg/day resulted in a statistically significant reduction in the longevity of males at 60 mg/kg/day and females at greater than or equal to 30 mg/kg/day. Target organs for neoplastic changes were the kidneys (renal tubule and renal pelvis), urinary bladder, urethra, preputial and clitoral gland, small intestine, parathyroid glands, adrenal glands and non-glandular stomach. Neoplastic lesions were not seen at: 30 mg/kg/day for the kidneys, urinary bladder, urethra, small intestine, parathyroid glands, adrenal glands and non-glandular stomach, and 15 mg/kg/day for the preputial and clitoral gland. The papilloma/carcinoma of the preputial/clitoral gland were noted at 30 and 60 mg/kg/day, representing approximately 0.5 to 4 or 0.3 to 2.4 times the human daily exposure (based on AUC) at 400 mg/day or 800 mg/day, respectively, and 0.4 to 3.0 times the daily exposure in children (based on AUC) at 340 mg/m². The renal tubule adenoma/carcinoma, renal pelvis transitional cell neoplasms, the urinary bladder and urethra transitional cell papillomas, the small intestine adenocarcinomas, the parathyroid glands adenomas, the benign and malignant medullary tumors of the adrenal glands and the non-glandular stomach papillomas/carcinomas were noted at 60 mg/kg/day. The relevance of these findings in the rat carcinogenicity study for humans is not known. Positive genotoxic effects were obtained for imatinib in an in vitro mammalian cell assay (Chinese hamster ovary) for clastogenicity (chromosome aberrations) in the presence of metabolic activation. Two intermediates of the manufacturing process, which are also present in the final product, are positive for mutagenesis in the Ames assay. One of these intermediates was also positive in the mouse lymphoma assay. Imatinib was not genotoxic when tested in an in vitro bacterial cell assay (Ames test), an in vitro mammalian cell assay (mouse lymphoma) and an in vivo rat micronucleus assay.
In a study of fertility, male rats were dosed for 70 days prior to mating and female rats were dosed 14 days prior to mating and through to gestational Day 6. Testicular and epididymal weights and percent motile sperm were decreased at 60 mg/kg, approximately three-fourths the maximum clinical dose of 800 mg/day based on body surface area. This was not seen at doses less than or equal to 20 mg/kg (one-fourth the maximum human dose of 800 mg). The fertility of male and female rats was not affected.
Fertility was not affected in the preclinical fertility and early embryonic development study although lower testes and epididymal weights as well as a reduced number of motile sperm were observed in the high dose males rats. In the preclinical pre-and postnatal study in rats, fertility in the first generation offspring was also not affected by imatinib mesylate.
Use In Specific Populations Pregnancy Risk SummaryGleevec can cause fetal harm when administered to a pregnant woman based on human and animal data. There are no clinical studies regarding use of Gleevec in pregnant women. There have been postmarket reports of spontaneous abortions and congenital anomalies from women who have been exposed to Gleevec during pregnancy. Reproductive studies in rats have demonstrated that imatinib mesylate induced teratogenicity and increased incidence of congenital abnormalities following prenatal exposure to imatinib mesylate at doses equal to the highest recommended human dose of 800 mg/day based on body surface area. Advise women to avoid pregnancy when taking Gleevec. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is not known; however, in the U.S. general population, the estimated background risk of major birth defects of clinically recognized pregnancies is 2-4% and of miscarriage is 15%-20%.
DataAnimal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of imatinib mesylate up to 100 mg/kg/day and 60 mg/kg/day, respectively, during the period of organogenesis.
In rats, imatinib mesylate was teratogenic at 100 mg/kg/day (approximately equal to the maximum human dose of 800 mg/day based on body surface area), the number of fetuses with encephalocoele and exencephaly was higher than historical control values and these findings were associated with missing or underdeveloped cranial bones. Lower mean fetal body weights were associated with retarded skeletal ossifications.
In rabbits, at doses 1.5 times higher than the maximum human dose of 800 mg/day based on body surface area, no effects on the reproductive parameters with respect to implantation sites, number of live fetuses, sex ratio or fetal weight were observed. The examinations of the fetuses did not reveal any drug related morphological changes.
In a pre-and postnatal development study in rats, pregnant rats received oral doses of imatinib mesylate during gestation (organogenesis) and lactation up to 45 mg/kg/day. Five animals developed a red vaginal discharge in the 45 mg/kg/day group on Days 14 or 15 of gestation, the significance of which is unknown since all females produced viable litters and none had increased post-implantation loss. Other maternal effects noted only at the dose of 45 mg/kg/day (approximately one-half the maximum human dose of 800 mg/day based on body surface area) included an increased numbers of stillborn pups and pups dying between postpartum Days 0 and 4. In the F1 offspring at this same dose level, mean body weights were reduced from birth until terminal sacrifice and the number of litters achieving criterion for preputial separation was slightly decreased. There were no other significant effects in developmental parameters or behavioral testing. F1 fertility was not affected but reproductive effects were noted at 45 mg/kg/day including an increased number of resorptions and a decreased number of viable fetuses. The NOEL for both maternal animals and the F1 generation was 15 mg/kg/day.
Lactation Risk SummaryImatinib and its active metabolite are excreted into human milk. Because of the potential for serious adverse reactions in breastfed infants from Gleevec, advise a lactating woman not to breastfeed during treatment and for 1 month after the last dose.
Human DataBased on data from 3 breastfeeding women taking Gleevec, the milk:plasma ratio is about 0.5 for imatinib and about 0.9 for the active metabolite. Considering the combined concentration of imatinib and active metabolite, a breastfed infant could receive up to 10% of the maternal therapeutic dose based on body weight.
Females And Males Of Reproductive Potential Pregnancy TestingHuman postmarketing reports and animal studies have shown Gleevec to be harmful to the developing fetus. Test pregnancy status in females with reproductive potential prior to the initiation of treatment with Gleevec.
ContraceptionFemales
Advise female patients of reproductive potential to use effective contraception (methods that result in less than 1 % pregnancy rates) when using Gleevec during treatment and for fourteen days after stopping treatment with Gleevec.
Infertility
The risk of infertility in females or males of reproductive potential has not been studied in humans. In a rat study, the fertility in males and females was not affected.
Pediatric UseThe safety and effectiveness of Gleevec have been demonstrated in pediatric patients with newly diagnosed Ph+ chronic phase CML and Ph+ ALL. There are no data in children under 1 year of age.
Geriatric UseIn the CML clinical studies, approximately 20% of patients were older than 65 years. In the study of patients with newly diagnosed CML, 6% of patients were older than 65 years. The frequency of edema was higher in patients older than 65 years as compared to younger patients; no other difference in the safety profile was observed. The efficacy of Gleevec was similar in older and younger patients.
In the unresectable or metastatic GIST study, 16% of patients were older than 65 years. No obvious differences in the safety or efficacy profile were noted in patients older than 65 years as compared to younger patients, but the small number of patients does not allow a formal analysis.
In the adjuvant GIST study, 221 patients (31%) were older than 65 years. No difference was observed in the safety profile in patients older than 65 years as compared to younger patients, with the exception of a higher frequency of edema. The efficacy of Gleevec was similar in patients older than 65 years and younger patients.
Hepatic ImpairmentThe effect of hepatic impairment on the pharmacokinetics of both imatinib and its major metabolite, CGP74588, was assessed in 84 patients with cancer with varying degrees of hepatic impairment at imatinib doses ranging from 100 mg to 800 mg.
Mild and moderate hepatic impairment do not influence exposure to imatinib and CGP74588. In patients with severe hepatic impairment, the imatinib Cmax and area under curve (AUC) increased by 63% and 45% and the CGP74588 Cmax and AUC increased by 56% and 55%, relative to patients with normal hepatic function. Reduce the dose by 25% for patients with severe hepatic impairment.
Table 16: Liver Function Classification
| Liver Function Test | Normal (n=14) |
Mild (n=30) |
Moderate (n=20) |
Severe (n=20) |
| Total Bilirubin | less than or equal to ULN | greater than 1.0-1.5 times the ULN | greater than 1.5-3 times the ULN | greater than 3-10 times the ULN |
| SGOT | less than or equal to ULN | greater than ULN (can be normal if Total Bilirubin is greater than ULN) | Any | Any |
| ULN=upper limit of normal for the institution |
The effect of renal impairment on the pharmacokinetics of imatinib was assessed in 59 patients with cancer and varying degrees of renal impairment at single and steady state imatinib doses ranging from 100 to 800 mg/day. The mean exposure to imatinib (dose normalized AUC) in patients with mild and moderate renal impairment increased 1.5-to 2-fold compared to patients with normal renal function. There are not sufficient data in patients with severe renal impairment. Dose reductions are necessary for patients with moderate and severe renal impairment.
Table 17: Renal Function
Classification
| Renal Dysfunction | Renal Function Tests |
| Mild | CrCL = 40-59 mL/min |
| Moderate | CrCL = 20-39 mL/min |
| Severe | CrCL = less than 20 mL/min |
| CrCL = Creatinine Clearance |
Dosage (Posology) and method of administration
Drug AdministrationThe prescribed dose should be administered orally, with a meal and a large glass of water. Doses of 400 mg or 600 mg should be administered once daily, whereas a dose of 800 mg should be administered as 400 mg twice a day.
For patients unable to swallow the film-coated tablets, the tablets may be dispersed in a glass of water or apple juice. The required number of tablets should be placed in the appropriate volume of beverage (approximately 50 mL for a 100 mg tablet, and 200 mL for a 400 mg tablet) and stirred with a spoon. The suspension should be administered immediately after complete disintegration of the tablet(s).
For daily dosing of 800 mg and above, dosing should be accomplished using the 400 mg tablet to reduce exposure to iron.
Treatment may be continued as long as there is no evidence of progressive disease or unacceptable toxicity.
Adult Patients With Ph+ CML CP, AP, Or BCThe recommended dose of Gleevec is 400 mg/day for adult patients in chronic phase CML and 600 mg/day for adult patients in accelerated phase or blast crisis.
In CML, a dose increase from 400 mg to 600 mg in adult patients with chronic phase disease, or from 600 mg to 800 mg (given as 400 mg twice daily) in adult patients in accelerated phase or blast crisis may be considered in the absence of severe adverse drug reaction and severe non-leukemia related neutropenia or thrombocytopenia in the following circumstances: disease progression (at any time), failure to achieve a satisfactory hematologic response after at least 3 months of treatment, failure to achieve a cytogenetic response after 6 to 12 months of treatment, or loss of a previously achieved hematologic or cytogenetic response.
Pediatric Patients With Ph+ CML CPThe recommended dose of Gleevec for children with newly diagnosed Ph+ CML is 340 mg/m²/day (not to exceed 600 mg). Gleevec treatment can be given as a once daily dose or the daily dose may be split into two– one portion dosed in the morning and one portion in the evening. There is no experience with Gleevec treatment in children under 1 year of age.
Adult Patients With Ph+ ALLThe recommended dose of Gleevec is 600 mg/day for adult patients with relapsed/refractory Ph+ ALL.
Pediatric Patients With Ph+ ALLThe recommended dose of Gleevec to be given in combination with chemotherapy to children with newly diagnosed Ph+ ALL is 340 mg/m²/day (not to exceed 600 mg). Gleevec treatment can be given as a once daily dose.
Adult Patients With MDS/MPDDetermine PDGFRb gene rearrangements status prior to initiating treatment. Information on FDA-approved tests for the detection of PDGFRb rearrangements is available at http://www.fda.gov/companiondiagnostics.
The recommended dose of Gleevec is 400 mg/day for adult patients with MDS/MPD.
Adult Patients With ASMDetermine D816V c-Kit mutation status prior to initiating treatment. Information on FDA-approved test for the detection of D816V c-Kit mutation is available at http://www.fda.gov/companiondiagnostics.
The recommended dose of Gleevec is 400 mg/day for adult patients with ASM without the D816V c-Kit mutation. If c-Kit mutational status is not known or unavailable, treatment with Gleevec 400 mg/day may be considered for patients with ASM not responding satisfactorily to other therapies. For patients with ASM associated with eosinophilia, a clonal hematological disease related to the fusion kinase FIP1L1-PDGFRα, a starting dose of 100 mg/day is recommended. Dose increase from 100 mg to 400 mg for these patients may be considered in the absence of adverse drug reactions if assessments demonstrate an insufficient response to therapy.
Adult Patients With HES/CELThe recommended dose of Gleevec is 400 mg/day for adult patients with HES/CEL. For HES/CEL patients with demonstrated FIP1L1-PDGFRα fusion kinase, a starting dose of 100 mg/day is recommended. Dose increase from 100 mg to 400 mg for these patients may be considered in the absence of adverse drug reactions if assessments demonstrate an insufficient response to therapy.
Adult Patients With DFSPThe recommended dose of Gleevec is 800 mg/day for adult patients with DFSP.
Adult Patients With Metastatic And/Or Unresectable GISTThe recommended dose of Gleevec is 400 mg/day for adult patients with unresectable and/or metastatic, malignant GIST. A dose increase up to 800 mg daily (given as 400 mg twice daily) may be considered, as clinically indicated, in patients showing clear signs or symptoms of disease progression at a lower dose and in the absence of severe adverse drug reactions.
Adult Patients With Adjuvant GISTThe recommended dose of Gleevec is 400 mg/day for the adjuvant treatment of adult patients following complete gross resection of GIST. In clinical trials, one year of Gleevec and three years of Gleevec were studied. In the patient population defined in Study 2, three years of Gleevec is recommended. The optimal treatment duration with Gleevec is not known.
Dose Modification Guidelines Concomitant Strong CYP3A4 InducersThe use of concomitant strong CYP3A4 inducers should be avoided (e.g., dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, rifampacin, phenobarbital). If patients must be coadministered a strong CYP3A4 inducer, based on pharmacokinetic studies, the dosage of Gleevec should be increased by at least 50%, and clinical response should be carefully monitored.
Hepatic ImpairmentPatients with mild and moderate hepatic impairment do not require a dose adjustment and should be treated per the recommended dose. A 25% decrease in the recommended dose should be used for patients with severe hepatic impairment.
Renal ImpairmentPatients with moderate renal impairment (CrCL=20–39 mL/min) should receive a 50% decrease in the recommended starting dose and future doses can be increased as tolerated. Doses greater than 600 mg are not recommended in patients with mild renal impairment (CrCL=40–59 mL/min). For patients with moderate renal impairment doses greater than 400 mg are not recommended.
Imatinib should be used with caution in patients with severe renal impairment. A dose of 100 mg/day was tolerated in two patients with severe renal impairment.
Dose Adjustment For Hepatotoxicity And Non-Hematologic Adverse ReactionsIf elevations in bilirubin greater than 3 times the institutional upper limit of normal (IULN) or in liver transaminases greater than 5 times the IULN occur, Gleevec should be withheld until bilirubin levels have returned to a less than 1.5 times the IULN and transaminase levels to less than 2.5 times the IULN. In adults, treatment with Gleevec may then be continued at a reduced daily dose (i.e., 400 mg to 300 mg, 600 mg to 400 mg or 800 mg to 600 mg). In children, daily doses can be reduced under the same circumstances from 340 mg/m²/day to 260 mg/m²/day.
If a severe non-hematologic adverse reaction develops (such as severe hepatotoxicity or severe fluid retention), Gleevec should be withheld until the event has resolved. Thereafter, treatment can be resumed as appropriate depending on the initial severity of the event.
Dose Adjustment For Hematologic Adverse ReactionsDose reduction or treatment interruptions for severe neutropenia and thrombocytopenia are recommended as indicated in Table 1.
Table 1: Dose Adjustments for Neutropenia and
Thrombocytopenia
| ASM associated with eosinophilia (starting dose 100 mg) | ANC1 less than 1.0 x 109/L and/or platelets less than 50 x 109/L |
|
| HES/CEL with FIP1L1-PDGFRα fusion kinase (starting dose 100 mg) | ANC less than 1.0 x 109/L and/or platelets less than 50 x 109/L |
|
| Chronic Phase CML (starting dose 400 mg) MDS/MPD, ASM and HES/CEL (starting dose 400 mg) GIST (starting dose 400 mg) |
ANC less than 1.0 x 109/L and/or platelets less than 50 x 109/L |
|
| Ph+ CML : Accelerated Phase and Blast Crisis (starting dose 600 mg) Ph+ ALL (starting dose 600 mg) | ANC less than 0.5 x 109/L and/or platelets less than 10 x 109/L |
|
| DFSP (starting dose 800 mg) | ANC less than 1.0 x 109/L and/or platelets less than 50 x 109/L |
|
| Pediatric newly diagnosed chronic phase CML (starting dose 340 mg/m²) | ANC less than 1.0 x 109/L and/or platelets less than 50 x 109/L |
|
| 1ANC = absolute neutrophil count |
Interaction with other medicinal products and other forms of interaction
Agents Inducing CYP3A MetabolismPretreatment of healthy volunteers with multiple doses of rifampin followed by a single dose of Gleevec, increased Gleevec oral-dose clearance by 3.8-fold, which significantly (p less than 0.05) decreased mean Cmax and AUC.
Similar findings were observed in patients receiving 400 to 1200 mg/day Gleevec concomitantly with enzyme-inducing anti-epileptic drugs (EIAED) (e.g., carbamazepine, oxcarbamazepine, phenytoin, fosphenytoin, phenobarbital, and primidone). The mean dose normalized AUC for imatinib in the patients receiving EIAED's decreased by 73% compared to patients not receiving EIAED.
Concomitant administration of Gleevec and St. John's Wort led to a 30% reduction in the AUC of imatinib.
Consider alternative therapeutic agents with less enzyme induction potential in patients when rifampin or other CYP3A4 inducers are indicated. Gleevec doses up to 1200 mg/day (600 mg twice daily) have been given to patients receiving concomitant strong CYP3A4 inducers.
Agents Inhibiting CYP3A MetabolismThere was a significant increase in exposure to imatinib (mean Cmax and AUC increased by 26% and 40%, respectively) in healthy subjects when Gleevec was coadministered with a single dose of ketoconazole (a CYP3A4 inhibitor). Caution is recommended when administering Gleevec with strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole). Grapefruit juice may also increase plasma concentrations of imatinib and should be avoided.
Interactions With Drugs Metabolized By CYP3A4Gleevec increases the mean Cmax and AUC of simvastatin (CYP3A4 substrate) 2-and 3.5-fold, respectively, suggesting an inhibition of the CYP3A4 by Gleevec. Particular caution is recommended when administering Gleevec with CYP3A4 substrates that have a narrow therapeutic window (e.g., alfentanil, cyclosporine, diergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus or tacrolimus).
Gleevec will increase plasma concentration of other CYP3A4 metabolized drugs (e.g., triazolobenzodiazepines, dihydropyridine calcium channel blockers, certain HMG-CoA reductase inhibitors, etc.).
Because warfarin is metabolized by CYP2C9 and CYP3A4, patients who require anticoagulation should receive low-molecular weight or standard heparin instead of warfarin.
Interactions With Drugs Metabolized By CYP2D6Gleevec increased the mean Cmax and AUC of metoprolol by approximately 23% suggesting that Gleevec has a weak inhibitory effect on CYP2D6-mediated metabolism. No dose adjustment is necessary, however, caution is recommended when administering Gleevec with CYP2D6 substrates that have a narrow therapeutic window.
Interactions With AcetaminophenIn vitro, Gleevec inhibits the acetaminophen O-glucuronidate pathway (Ki 58.5 μM). Coadministration of Gleevec (400 mg/day for 8 days) with acetaminophen (1000 mg single dose on day 8) in patients with CML did not result in any changes in the pharmacokinetics of acetaminophen. Gleevec pharmacokinetics were not altered in the presence of single-dose acetaminophen. There is no pharmacokinetic or safety data on the concomitant use of Gleevec at doses greater than 400 mg/day or the chronic use of concomitant acetaminophen and Gleevec.