Chantix
Overdose
In case of overdose, standard supportive measures should be instituted as required.
Varenicline has been shown to be dialyzed in patients with end-stage renal disease , however, there is no experience in dialysis following overdose.
Contraindications
CHANTIX is contraindicated in patients with a known history of serious hypersensitivity reactions or skin reactions to CHANTIX.
Undesirable effects
The following serious adverse reactions were reported in postmarketing experience and are discussed in greater detail in other sections of the labeling:
- Neuropsychiatric Adverse Events including Suicidality
- Seizures
- Interaction with alcohol
- Accidental injury
- Cardiovascular events
- Somnambulism
- Angioedema and hypersensitivity reactions
- Serious skin reactions
In the placebo-controlled premarketing studies, the most common adverse events associated with CHANTIX (>5% and twice the rate seen in placebo-treated patients) were nausea, abnormal (vivid, unusual, or strange) dreams, constipation, flatulence, and vomiting.
The treatment discontinuation rate due to adverse events in patients dosed with 1 mg twice daily was 12% for CHANTIX, compared to 10% for placebo in studies of three months' treatment. In this group, the discontinuation rates that are higher than placebo for the most common adverse events in CHANTIX-treated patients were as follows: nausea (3% vs. 0.5% for placebo), insomnia (1.2% vs. 1.1% for placebo), and abnormal dreams (0.3% vs. 0.2% for placebo).
Smoking cessation, with or without treatment, is associated with nicotine withdrawal symptoms and has also been associated with the exacerbation of underlying psychiatric illness.
Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, the adverse reactions rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
During the premarketing development of CHANTIX, over 4500 subjects were exposed to CHANTIX, with over 450 treated for at least 24 weeks and approximately 100 for a year. Most study participants were treated for 12 weeks or less.
The most common adverse event associated with CHANTIX treatment is nausea, occurring in 30% of patients treated at the recommended dose, compared with 10% in patients taking a comparable placebo regimen.
Table 3 shows the adverse events for CHANTIX and placebo in the 12- week fixed dose premarketing studies with titration in the first week [Studies 2 (titrated arm only), 4, and 5]. Adverse events were categorized using the Medical Dictionary for Regulatory Activities (MedDRA, Version 7.1).
MedDRA High Level Group Terms (HLGT) reported in ≥ 5% of patients in the CHANTIX 1 mg twice daily dose group, and more commonly than in the placebo group, are listed, along with subordinate Preferred Terms (PT) reported in ≥ 1% of CHANTIX patients (and at least 0.5% more frequent than placebo). Closely related Preferred Terms such as 'Insomnia', 'Initial insomnia', 'Middle insomnia', 'Early morning awakening' were grouped, but individual patients reporting two or more grouped events are only counted once.
Table 3: Common Treatment Emergent AEs (%) in the
Fixed-Dose, Placebo- Controlled Studies (HLGTs ≥ 5% of patients in the 1
mg BID CHANTIX Group and more commonly than Placebo and PT ≥ 1% in the 1
mg BID CHANTIX Group, and 1 mg BID CHANTIX at least 0.5% more than Placebo)
| SYSTEM ORGAN CLASS High Level Group Term Preferred Term | CHANTIX 0.5 mg BID N=129 |
CHANTIX 1 mg BID N=821 |
Placebo N=805 |
| GASTROINTESTINAL (GI) | |||
| GI Signs and Symptoms | |||
| Nausea | 16 | 30 | 10 |
| Abdominal Pain * | 5 | 7 | 5 |
| Flatulence | 9 | 6 | 3 |
| Dyspepsia | 5 | 5 | 3 |
| Vomiting | 1 | 5 | 2 |
| GI Motility/Defecation Conditions | |||
| Constipation | 5 | 8 | 3 |
| Gastroesophageal reflux disease | 1 | 1 | 0 |
| Salivary Gland Conditions | |||
| Dry mouth | 4 | 6 | 4 |
| PSYCHIATRIC DISORDERS | |||
| Sleep Disorder/Disturbances | |||
| Insomnia† | 19 | 18 | 13 |
| Abnormal dreams | 9 | 13 | 5 |
| Sleep disorder | 2 | 5 | 3 |
| Nightmare | 2 | 1 | 0 |
| NERVOUS SYSTEM | |||
| Headaches | |||
| Headache | 19 | 15 | 13 |
| Neurological Disorders NEC | |||
| Dysgeusia | 8 | 5 | 4 |
| Somnolence | 3 | 3 | 2 |
| Lethargy | 2 | 1 | 0 |
| GENERAL DISORDERS | |||
| General Disorders NEC | |||
| Fatigue/Malaise/Asthenia | 4 | 7 | 6 |
| RESPIR/THORACIC/MEDIAST | |||
| Respiratory Disorders NEC | |||
| Rhinorrhea | 0 | 1 | 0 |
| Dyspnea | 2 | 1 | 1 |
| Upper Respiratory Tract Disorder | 7 | 5 | 4 |
| SKIN/SUBCUTANEOUS TISSUE | |||
| Epidermal and Dermal Conditions | |||
| Rash | 1 | 3 | 2 |
| Pruritis | 0 | 1 | 1 |
| METABOLISM & NUTRITION | |||
| Appetite/General mNutrition Disorders | |||
| Increased appetite | 4 | 3 | 2 |
| Decreased appetite/Anorexia | 1 | 2 | 1 |
| *Includes PTs Abdominal (pain, pain upper, pain lower,
discomfort, tenderness, distension) and Stomach discomfort †Includes PTs Insomnia/Initial insomnia/Middle insomnia/Early morning awakening |
|||
The overall pattern and frequency of adverse events during the longer-term premarketing trials was similar to those described in Table 3, though several of the most common events were reported by a greater proportion of patients with long-term use (e.g., nausea was reported in 40% of patients treated with CHANTIX 1 mg twice daily in a one year study, compared to 8% of placebo-treated patients).
Following is a list of treatment-emergent adverse events reported by patients treated with CHANTIX during all premarketing clinical trials and updated based on pooled data from 18 placebo-controlled preand post-marketing studies, including approximately 5,000 patients treated with varenicline. Adverse events were categorized using MedDRA, Version 16.0. The listing does not include those events already listed in the previous tables or elsewhere in labeling, those events for which a drug cause was remote, those events which were so general as to be uninformative, and those events reported only once which did not have a substantial probability of being acutely life-threatening.
Blood and Lymphatic System Disorders. Infrequent: anemia, lymphadenopathy. Rare: leukocytosis, splenomegaly, thrombocytopenia.
Cardiac Disorders. Infrequent: angina pectoris, myocardial infarction, palpitations, tachycardia. Rare: acute coronary syndrome, arrhythmia, atrial fibrillation, bradycardia, cardiac flutter, cor pulmonale, coronary artery disease, ventricular extrasystoles.
Ear and Labyrinth Disorders. Infrequent: tinnitus, vertigo. Rare: deafness, Meniere's disease.
Endocrine Disorders. Infrequent: thyroid gland disorders.
Eye Disorders. Infrequent: conjunctivitis, eye irritation, eye pain, vision blurred, visual impairment.
Rare: blindness transient, cataract subcapsular, dry eye, night blindness, ocular vascular disorder, photophobia, vitreous floaters.
Gastrointestinal Disorders. Frequent: diarrhea, toothache. Infrequent: dysphagia, eructation, gastritis, gastrointestinal hemorrhage, mouth ulceration. Rare: enterocolitis, esophagitis, gastric ulcer, intestinal obstruction, pancreatitis acute.
General Disorders and Administration Site Conditions. Frequent: chest pain. Infrequent: chest discomfort, chills, edema, influenza-like illness, pyrexia.
Hepatobiliary Disorders. Rare: gall bladder disorder.
Investigations. Frequent: liver function test abnormal, weight increased. Infrequent: electrocardiogram abnormal. Rare: muscle enzyme increased, urine analysis abnormal.
Metabolism and Nutrition Disorders. Infrequent: diabetes mellitus, hypoglycemia. Rare: hyperlipidemia, hypokalemia.
Musculoskeletal and Connective Tissue Disorders. Frequent: arthralgia, back pain, myalgia. Infrequent: arthritis, muscle cramp, musculoskeletal pain. Rare: myositis, osteoporosis.
Nervous System Disorders. Frequent: disturbance in attention, dizziness. Infrequent: amnesia, convulsion, migraine, parosmia, syncope, tremor. Rare: balance disorder, cerebrovascular accident, dysarthria, mental impairment, multiple sclerosis, VII th  nerve paralysis, nystagmus, psychomotor hyperactivity, psychomotor skills impaired, restless legs syndrome, sensory disturbance, transient ischemic attack, visual field defect.
Psychiatric Disorders. Infrequent: dissociation, libido decreased, mood swings, thinking abnormal. Rare: bradyphrenia, disorientation, euphoric mood.
Renal and Urinary Disorders. Infrequent: nocturia, pollakiuria, urine abnormality. Rare: nephrolithiasis, polyuria, renal failure acute, urethral syndrome, urinary retention.
Reproductive System and Breast Disorders. Frequent: menstrual disorder. Infrequent: erectile dysfunction. Rare: sexual dysfunction.
Respiratory, Thoracic and Mediastinal Disorders. Frequent: respiratory disorders. Infrequent: asthma, epistaxis, rhinitis allergic, upper respiratory tract inflammation. Rare: pleurisy, pulmonary embolism.
Skin and Subcutaneous Tissue Disorders. Infrequent: acne, dry skin, eczema, erythema, hyperhidrosis, urticaria. Rare: photosensitivity reaction, psoriasis.
Vascular Disorders. Infrequent: hot flush. Rare: thrombosis.
CHANTIX has also been studied in postmarketing trials including (1) a trial conducted in patients with chronic obstructive pulmonary disease (COPD), (2) a trial conducted in generally healthy patients (similar to those in the premarketing studies) in which they were allowed to select a quit date between days 8 and 35 of treatment (“alternative quit date instruction trial”), (3) a trial conducted in patients who did not succeed in stopping smoking during prior CHANTIX therapy, or who relapsed after treatment (“re-treatment trial”), (4) a trial conducted in patients with stable cardiovascular disease, (5) a trial conducted in patients with stable schizophrenia or schizoaffective disorder, (6) a trial conducted in patients with major depressive disorder, (7) a postmarketing neuropsychiatric safety outcome trial in patients without or with a history of psychiatric disorder, and (8) a trial in patients who were not able or willing to quit abruptly and who were instructed to quit gradually (“gradual approach to quitting smoking trial”).
Adverse events in the trial of patients with COPD, in the alternative quit date instruction trial, and in the gradual approach to quitting smoking trial were similar to those observed in premarketing studies. In the re-treatment trial, the profile of common adverse events was similar to that previously reported, but, in addition, varenicline-treated patients also commonly reported diarrhea (6% vs. 4% in placebo-treated patients), depressed mood disorders and disturbances (6% vs. 1%), and other mood disorders and disturbances (5% vs. 2%).
In the trial of patients with stable cardiovascular disease, more types and a greater number of cardiovascular events were reported compared to premarketing studies. Treatment-emergent (ontreatment or 30 days after treatment) cardiovascular events reported with a frequency ≥ 1% in either treatment group in this study were angina pectoris (3.7% and 2.0% for varenicline and placebo, respectively), chest pain (2.5% vs. 2.3%), peripheral edema (2.0% vs. 1.1%), hypertension (1.4% vs. 2.6%), and palpitations (0.6 % vs. 1.1%). Deaths and serious cardiovascular events occurring over the 52 weeks of the study (treatment emergent and non-treatment emergent) were adjudicated by a blinded, independent committee. The following treatment-emergent adjudicated events occurred with a frequency ≥ 1% in either treatment group: nonfatal MI (1.1% vs. 0.3% for varenicline and placebo, respectively), and hospitalization for angina pectoris (0.6% vs. 1.1%). During non-treatment follow-up to 52 weeks, the adjudicated events included need for coronary revascularization (2.0% vs. 0.6%), hospitalization for angina pectoris (1.7% vs. 1.1%), and new diagnosis of peripheral vascular disease (PVD) or admission for a PVD procedure (1.4% vs. 0.6%). Some of the patients requiring coronary revascularization underwent the procedure as part of management of nonfatal MI and hospitalization for angina. Cardiovascular death occurred in 0.3% of patients in the varenicline arm and 0.6% of patients in the placebo arm over the course of the 52-week study.
In the trial of patients with stable schizophrenia or schizoaffective disorder, 128 smokers on antipsychotic medication were randomized 2:1 to varenicline (1 mg twice daily) or placebo for 12 weeks with 12-week non-drug follow-up. The most common adverse events in patients taking varenicline were nausea (24% vs. 14.0% on placebo), headache (11% vs. 19% on placebo) and vomiting (11% vs. 9% on placebo). Among reported neuropsychiatric adverse events, insomnia was the only event that occurred in either treatment group in ≥5% of subjects at a rate higher in the varenicline group than in placebo (10% vs. 5%). These common and neuropsychiatric adverse events occurred on treatment or within 30 days after the last dose of study drug. There was no consistent worsening of schizophrenia in either treatment group as measured by the Positive and Negative Syndrome Scale. There were no overall changes in extra-pyramidal signs, as measured by the Simpson-Angus Rating Scale. The Columbia-Suicide Severity Rating Scale was administered at baseline and at clinic visits during the treatment and non-treatment follow-up phases. Over half of the patients had a lifetime history of suicidal behavior and/or ideation (62% on varenicline vs. 51% on placebo), but at baseline, no patients in the varenicline group reported suicidal behavior and/or ideation vs. one patient in the placebo group (2%). Suicidal behavior and/or ideation were reported in 11% of the varenicline-treated and 9% of the placebo-treated patients during the treatment phase. During the post-treatment phase, suicidal behavior and/or ideation were reported in 11% of patients in the varenicline group and 5% of patients in the placebo group. Many of the patients reporting suicidal behavior and ideation in the follow-up phase had not reported such experiences in the treatment phase. However, no new suicidal ideation or behavior emerged in either treatment group shortly (within one week) after treatment discontinuation (a phenomenon noted in postmarketing reporting). There were no completed suicides. There was one suicide attempt in a varenicline-treated patient. The limited data available from this single smoking cessation study are not sufficient to allow conclusions to be drawn.
In the trial of patients with major depressive disorder, the most common adverse events (≥ 10%) in subjects taking varenicline were nausea (27% vs. 10% on placebo), headache (17 vs. 11%), abnormal dreams (11% vs. 8%), insomnia (11% vs. 5%) and irritability (11% vs. 8%). Additionally, the following psychiatric AEs were reported in ≥ 2% of patients in either treatment group (varenicline or placebo, respectively): anxiety (7% vs. 9%), agitation (7% vs. 4%), depressed mood disorders and disturbances (11% vs. 9%), tension (4% vs. 3%), hostility (2% vs. 0.4%) and restlessness (2% vs. 2%). Patients treated with varenicline were more likely than patients treated with placebo to report one of various events related to hostility and aggression (3% vs. 1%). Psychiatric scales showed no differences between the varenicline and placebo groups and no overall worsening of depression during the study in either treatment group. The percentage of subjects with suicidal ideation and/or behavior was similar between the varenicline and placebo groups during treatment (6% and 8%, respectively) and the nontreatment follow-up (6% and 6%, respectively). There was one event of intentional self-injury/possible suicide attempt during treatment (Day 73) in a subject in the placebo group. Suicide could not be ruled out in one subject who died by an overdose of illicit drugs 76 days after last dose of study drug in the varenicline group.
In the trial of patients without or with a history of psychiatric disorder, the most common adverse events in subjects treated with varenicline were similar to those observed in premarketing studies. Adverse events reported in ≥ 10% of subjects treated with varenicline in the entire study population were nausea (25% vs. 7% on placebo) and headache (12% vs. 10% on placebo). Additionally, the following psychiatric adverse events were reported in ≥ 2% of patients in either treatment group (varenicline vs. placebo) by cohort. For the non-psychiatric cohort, these adverse events were abnormal dreams (8% vs. 4%), agitation (3% vs. 3%), anxiety (5% vs. 6%), depressed mood (3% vs. 3%), insomnia (10% vs. 7%), irritability (3% vs. 4%), sleep disorder (3% vs. 2%). For the psychiatric cohort, these adverse events were abnormal dreams (12% vs. 5%), agitation (5% vs. 4%), anxiety (8% vs. 6%), depressed mood (5% vs. 5%), depression (5% vs. 5%), insomnia (9% vs. 7%), irritability (5% vs. 7%), nervousness (2% vs. 3%), sleep disorder (3% vs. 2%).
Postmarketing ExperienceThe following adverse events have been reported during post-approval use of CHANTIX. Because these events are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
There have been reports of depression, mania, psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, anxiety, and panic, as well as suicidal ideation, suicide attempt, and completed suicide in patients attempting to quit smoking while taking CHANTIX.
There have been postmarketing reports of new or worsening seizures in patients treated with CHANTIX.
There have been postmarketing reports of patients experiencing increased intoxicating effects of alcohol while taking CHANTIX. Some reported neuropsychiatric events, including unusual and sometimes aggressive behavior.
There have been reports of hypersensitivity reactions, including angioedema.
There have also been reports of serious skin reactions, including Stevens-Johnson Syndrome and erythema multiforme, in patients taking CHANTIX. There have been reports of myocardial infarction (MI) and cerebrovascular accident (CVA) including ischemic and hemorrhagic events in patients taking CHANTIX. In the majority of the reported cases, patients had pre-existing cardiovascular disease and/or other risk factors. Although smoking is a risk factor for MI and CVA, based on temporal relationship between medication use and events, a contributory role of varenicline cannot be ruled out.
There have been reports of hyperglycemia in patients following initiation of CHANTIX. There have been reports of somnambulism, some resulting in harmful behavior to self, others, or property in patients treated with CHANTIX.
Therapeutic indications
CHANTIX is indicated for use as an aid to smoking cessation treatment.
Pharmacokinetic properties
AbsorptionMaximum plasma concentrations of varenicline occur typically within 3-4 hours after oral administration. Following administration of multiple oral doses of varenicline, steady-state conditions were reached within 4 days. Over the recommended dosing range, varenicline exhibits linear pharmacokinetics after single or repeated doses.
In a mass balance study, absorption of varenicline was virtually complete after oral administration and systemic availability was ~90%.
Food Effect
Oral bioavailability of varenicline is unaffected by food or time-of-day dosing.
DistributionPlasma protein binding of varenicline is low (≤ 20%) and independent of both age and renal function.
EliminationThe elimination half-life of varenicline is approximately 24 hours.
Metabolism
Varenicline undergoes minimal metabolism, with 92% excreted unchanged in the urine.
Excretion
Renal elimination of varenicline is primarily through glomerular filtration along with active tubular secretion possibly via the organic cation transporter, OCT2.
Date of revision of the text
Dec 20 16Name of the medicinal product
ChantixFertility, pregnancy and lactation
Risk SummaryAvailable human data on the use of CHANTIX in pregnant women are not sufficient to inform a drug associated risk. Smoking during pregnancy is associated with maternal, fetal, and neonatal risks. In animal studies, varenicline did not result in major malformations but caused decreased fetal weights in rabbits when dosed during organogenesis at exposures equivalent to 50 times the exposure at the maximum recommended human dose (MRHD). Additionally, administration of varenicline to pregnant rats during organogenesis through lactation produced developmental toxicity in offspring at maternal exposures equivalent to 36 times human exposure at the MRHD.
The estimated background risk of oral clefts is increased by approximately 30% in infants of women who smoke during pregnancy, compared to pregnant women who do not smoke. The background risk of other major birth defects and miscarriage for the indicated population are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Clinical ConsiderationsDisease-Associated Maternal And/Or Embryo/Fetal Risk
Smoking during pregnancy causes increased risks of orofacial clefts, premature rupture of membranes, placenta previa, placental abruption, ectopic pregnancy, fetal growth restriction and low birth weight, stillbirth, preterm delivery and shortened gestation, neonatal death, sudden infant death syndrome and reduction of lung function in infants. It is not known whether quitting smoking with CHANTIX during pregnancy reduces these risks.
DataAnimal Data
Pregnant rats and rabbits received varenicline succinate during organogenesis at oral doses up to 15 and 30 mg/kg/day, respectively. While no fetal structural abnormalities occurred in either species, maternal toxicity, characterized by reduced body weight gain, and reduced fetal weights occurred in rabbits at the highest dose (exposures 50 times the human exposure at the MRHD of 1 mg twice daily based on AUC). Fetal weight reduction did not occur in rabbits at exposures 23 times the human exposure at the MRHD based on AUC.
In a pre- and postnatal development study, pregnant rats received up to 15 mg/kg/day of oral varenicline succinate from organogenesis through lactation. Maternal toxicity, characterized by a decrease in body weight gain was observed at 15 mg/kg/day (36 times the human exposure at the MRHD based on AUC). However, decreased fertility and increased auditory startle response occurred in offspring at the highest maternal dose of 15 mg/kg/day.
Qualitative and quantitative composition
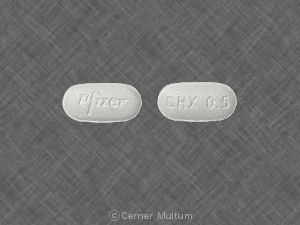
Dosage Forms And StrengthsCapsular, biconvex tablets: 0.5 mg (white to off-white, debossed with “Pfizer” on one side and “CHX 0.5” on the other side) and 1 mg (light blue, debossed with “Pfizer” on one side and “CHX 1.0” on the other side).
Storage And HandlingCHANTIX is supplied for oral administration in two strengths: a 0.5 mg capsular biconvex, white to off-white, film-coated tablet debossed with “Pfizer” on one side and “CHX 0.5” on the other side and a 1 mg capsular biconvex, light blue film-coated tablet debossed with “Pfizer” on one side and “CHX 1.0” on the other side. CHANTIX is supplied in the following package configurations:
| Description | NDC | |
| Packs | Starting 2 week card: 0.5 mg x 11 tablets and 1 mg x 14 tablets | NDC 0069-0471-01 |
| Continuing 2 week card: 1 mg x 28 tablets | NDC 0069-0469-11 | |
| Starting 4-week card: 0.5 mg x 11 tablets and 1 mg x 42 tablets |
NDC 0069-0471-03 | |
| Continuing 4-week card: 1 mg x 56 tablets | NDC 0069-0469-03 | |
| Starting Month Box: 0.5 mg x 11 tablets and 1 mg x 42 tablets |
NDC 0069-0471-02; NDC 0069-0471-03 |
|
| Continuing Month Box: 1 mg x 56 tablets | NDC 0069-0469-12; NDC 0069-0469-03 |
|
| Bottles | 0.5 mg - bottle of 56 | NDC 0069-0468-56 |
| 1 mg - bottle of 56 | NDC 0069-0469-56 |
Store at 25°C (77°F); excursions permitted to 15–30°C (59–86°F) (see USP Controlled Room Temperature).
Distributed by: Pfizer Labs, Division of Pfizer Inc, NY, NY 10017. Revised: Dec 20 16
Special warnings and precautions for use
WARNINGSIncluded as part of the PRECAUTIONS section.
PRECAUTIONS Neuropsychiatric Adverse Events Including SuicidalitySerious neuropsychiatric adverse events have been reported in patients being treated with CHANTIX. These postmarketing reports have included changes in mood (including depression and mania), psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, agitation, anxiety, and panic, as well as suicidal ideation, suicide attempt, and completed suicide. Some patients who stopped smoking may have been experiencing symptoms of nicotine withdrawal, including depressed mood. Depression, rarely including suicidal ideation, has been reported in smokers undergoing a smoking cessation attempt without medication. However, some of these adverse events occurred in patients taking CHANTIX who continued to smoke.
Neuropsychiatric adverse events occurred in patients without and with pre-existing psychiatric disease; some patients experienced worsening of their psychiatric illnesses. Some neuropsychiatric adverse events, including unusual and sometimes aggressive behavior directed to oneself or others, may have been worsened by concomitant use of alcohol. Observe patients for the occurrence of neuropsychiatric adverse events. Advise patients and caregivers that the patient should stop taking CHANTIX and contact a healthcare provider immediately if agitation, depressed mood, or changes in behavior or thinking that are not typical for the patient are observed, or if the patient develops suicidal ideation or suicidal behavior. The healthcare provider should evaluate the severity of the symptoms and the extent to which the patient is benefiting from treatment, and consider options including dose reduction, continued treatment under closer monitoring, or discontinuing treatment. In many postmarketing cases, resolution of symptoms after discontinuation of CHANTIX was reported. However, the symptoms persisted in some cases; therefore, ongoing monitoring and supportive care should be provided until symptoms resolve.
The neuropsychiatric safety of CHANTIX was evaluated in a randomized, double-blind, active and placebo-controlled study that included patients without a history of psychiatric disorder (nonpsychiatric cohort, N=3912) and patients with a history of psychiatric disorder (psychiatric cohort, N=4003). In the non-psychiatric cohort, CHANTIX was not associated with an increased incidence of clinically significant neuropsychiatric adverse events in a composite endpoint comprising anxiety, depression, feeling abnormal, hostility, agitation, aggression, delusions, hallucinations, homicidal ideation, mania, panic, and irritability. In the psychiatric cohort, there were more events reported in each treatment group compared to the non-psychiatric cohort, and the incidence of events in the composite endpoint was higher for each of the active treatments compared to placebo: Risk Differences (RDs) (95%CI) vs. placebo were 2.7% (-0.05, 5.4) for CHANTIX, 2.2% (-0.5, 4.9) for bupropion, and 0.4% (-2.2, 3.0) for transdermal nicotine. In the non-psychiatric cohort, neuropsychiatric adverse events of a serious nature were reported in 0.1% of CHANTIX-treated patients and 0.4% of placebo-treated patients. In the psychiatric cohort, neuropsychiatric events of a serious nature were reported in 0.6% of CHANTIX-treated patients, with 0.5% involving psychiatric hospitalization. In placebo-treated patients, serious neuropsychiatric events occurred in 0.6%, with 0.2% requiring psychiatric hospitalization.
SeizuresDuring clinical trials and the post marketing experience, there have been reports of seizures in patients treated with CHANTIX. Some patients had no history of seizures, whereas others had a history of seizure disorder that was remote or well-controlled. In most cases, the seizure occurred within the first month of therapy. Weigh this potential risk against the potential benefits before prescribing CHANTIX in patients with a history of seizures or other factors that can lower the seizure threshold. Advise patients to discontinue CHANTIX and contact a healthcare provider immediately if they experience a seizure while on treatment.
Interaction With AlcoholThere have been postmarketing reports of patients experiencing increased intoxicating effects of alcohol while taking CHANTIX. Some cases described unusual and sometimes aggressive behavior, and were often accompanied by amnesia for the events. Advise patients to reduce the amount of alcohol they consume while taking CHANTIX until they know whether CHANTIX affects their tolerance for alcohol.
Accidental InjuryThere have been postmarketing reports of traffic accidents, near-miss incidents in traffic, or other accidental injuries in patients taking CHANTIX. In some cases, the patients reported somnolence, dizziness, loss of consciousness or difficulty concentrating that resulted in impairment, or concern about potential impairment, in driving or operating machinery. Advise patients to use caution driving or operating machinery or engaging in other potentially hazardous activities until they know how CHANTIX may affect them.
Cardiovascular EventsIn a placebo-controlled clinical trial of CHANTIX administered to patients with stable cardiovascular disease, with approximately 350 patients per treatment arm, all-cause and cardiovascular mortality was lower in patients treated with CHANTIX, but certain nonfatal cardiovascular events occurred more frequently in patients treated with CHANTIX than in patients treated with placebo. Table 1 below shows the incidence of deaths and of selected nonfatal serious cardiovascular events occurring more frequently in the CHANTIX arm compared to the placebo arm. These events were adjudicated by an independent blinded committee. Nonfatal serious cardiovascular events not listed occurred at the same incidence or more commonly in the placebo arm. Patients with more than one cardiovascular event of the same type are counted only once per row. Some of the patients requiring coronary revascularization underwent the procedure as part of management of nonfatal MI and hospitalization for angina.
Table 1: Mortality and Adjudicated Nonfatal Serious
Cardiovascular Events in the Placebo-Controlled CHANTIX Trial in Patients with
Stable Cardiovascular Disease
| Mortality and Cardiovascular Events | CHANTIX (N=353) n (%) |
Placebo (N=350) n (%) |
| Mortality (Cardiovascular & All-cause up to 52 wks) | ||
| Cardiovascular death | 1 (0.3) | 2 (0.6) |
| All-cause mortality | 2 (0.6) | 5 (1.4) |
| Nonfatal Cardiovascular Events (rate on CHANTIX > Placebo) | ||
| Up to 30 days after treatment | ||
| Nonfatal myocardial infarction | 4 (1.1) | 1 (0.3) |
| Nonfatal Stroke | 2(0.6) | 0 (0) |
| Beyond 30 days after treatment & up to 52 weeks | ||
| Nonfatal myocardial infarction | 3(0.8) | 2 (0.6) |
| Need for coronary revascularization | 7 (2.0) | 2 (0.6) |
| Hospitalization for angina pectoris | 6 (1.7) | 4 (1.1) |
| Transient ischemia attack | 1 (0.3) | 0 (0) |
| New diagnosis of peripheral vascular disease (PVD) or admission for a PVD procedure | 5 (1.4) | 2 (0.6) |
A meta-analysis of 15 clinical trials of ≥ 12 weeks treatment duration, including 7002 patients (4190 CHANTIX, 2812 placebo), was conducted to systematically assess the cardiovascular safety of CHANTIX. The study in patients with stable cardiovascular disease described above was included in the meta-analysis. There were lower rates of all-cause mortality (CHANTIX 6 [0.14%]; placebo 7 [0.25%]) and cardiovascular mortality (CHANTIX 2 [0.05%]; placebo 2 [0.07%]) in the CHANTIX arms compared with the placebo arms in the meta-analysis.
The key cardiovascular safety analysis included occurrence and timing of a composite endpoint of Major Adverse Cardiovascular Events (MACE), defined as cardiovascular death, nonfatal MI, and nonfatal stroke. These events included in the endpoint were adjudicated by a blinded, independent committee. Overall, a small number of MACE occurred in the trials included in the meta-analysis, as described in Table 2. These events occurred primarily in patients with known cardiovascular disease.
Table 2: Number of MACE cases, Hazard Ratio and Rate
Difference in a Meta-Analysis of 15 Clinical Trials Comparing CHANTIX to
Placebo*
| CHANTIX N=4190 |
Placebo N=2812 |
|
| MACE cases, n (%) | 13 (0.31%) |
6 (0.21%) |
| Patient-years of exposure | 1316 | 839 |
| Hazard Ratio (95% CI) | ||
| 1.95 (0.79, 4.82) |
||
| Rate Difference per 1,000 patient-years (95% CI) | ||
| 6.30 (-2.40, 15.10) |
||
| *Includes MACE occurring up to 30 days post-treatment. | ||
The meta-analysis showed that exposure to CHANTIX resulted in a hazard ratio for MACE of 1.95 (95% confidence interval from 0.79 to 4.82) for patients up to 30 days after treatment; this is equivalent to an estimated increase of 6.3 MACE events per 1,000 patient-years of exposure. The meta-analysis showed higher rates of CV endpoints in patients on CHANTIX relative to placebo across different time frames and pre-specified sensitivity analyses, including various study groupings and CV outcomes. Although these findings were not statistically significant they were consistent. Because the number of events was small overall, the power for finding a statistically significant difference in a signal of this magnitude is low.
CHANTIX was not studied in patients with unstable cardiovascular disease or cardiovascular events occurring within two months before screening. Patients should be advised to notify a healthcare provider of new or worsening symptoms of cardiovascular disease. The risks of CHANTIX should be weighed against the benefits of its use in smokers with cardiovascular disease. Smoking is an independent and major risk factor for cardiovascular disease. CHANTIX has been demonstrated to increase the likelihood of abstinence from smoking for as long as one year compared to treatment with placebo.
SomnambulismCases of somnambulism have been reported in patients taking CHANTIX. Some cases described harmful behavior to self, others, or property. Instruct patients to discontinue CHANTIX and notify their healthcare provider if they experience somnambulism.
Angioedema And Hypersensitivity ReactionsThere have been postmarketing reports of hypersensitivity reactions including angioedema in patients treated with CHANTIX. Clinical signs included swelling of the face, mouth (tongue, lips, and gums), extremities, and neck (throat and larynx). There were infrequent reports of life-threatening angioedema requiring emergent medical attention due to respiratory compromise. Instruct patients to discontinue CHANTIX and immediately seek medical care if they experience these symptoms.
Serious Skin ReactionsThere have been postmarketing reports of rare but serious skin reactions, including Stevens-Johnson Syndrome and erythema multiforme, in patients using CHANTIX. As these skin reactions can be life-threatening, instruct patients to stop taking CHANTIX and contact a healthcare provider immediately at the first appearance of a skin rash with mucosal lesions or any other signs of hypersensitivity.
NauseaNausea was the most common adverse reaction reported with CHANTIX treatment. Nausea was generally described as mild or moderate and often transient; however, for some patients, it was persistent over several months. The incidence of nausea was dose-dependent. Initial dose-titration was beneficial in reducing the occurrence of nausea. For patients treated to the maximum recommended dose of 1 mg twice daily following initial dosage titration, the incidence of nausea was 30% compared with 10% in patients taking a comparable placebo regimen. In patients taking CHANTIX 0.5 mg twice daily following initial titration, the incidence was 16% compared with 11% for placebo. Approximately 3% of patients treated with CHANTIX 1 mg twice daily in studies involving 12 weeks of treatment discontinued treatment prematurely because of nausea. For patients with intolerable nausea, a dose reduction should be considered.
Patient Counseling InformationSee FDA-approved patient labeling (Medication Guide)
Initiate Treatment And Continue To Attempt To Quit If LapseInstruct patients to set a date to quit smoking and to initiate CHANTIX treatment one week before the quit date. Alternatively, the patient can begin CHANTIX dosing and then set a date to quit smoking between days 8 and 35 of treatment. Encourage patients to continue to attempt to quit if they have early lapses after quit day.
For patients who are sure that they are not able or willing to quit abruptly, a gradual approach to quitting smoking with CHANTIX may be considered. Patients should begin CHANTIX dosing and reduce smoking during the first 12 weeks of treatment, then quit by the end of that period and continue treatment for an additional 12 weeks for a total of 24 weeks.
Encourage patients who are motivated to quit and who did not succeed in stopping smoking during prior CHANTIX therapy for reasons other than intolerability due to adverse events, or who relapsed after treatment to make another attempt with CHANTIX once factors contributing to the failed attempt have been identified and addressed.
How To TakeAdvise patients that CHANTIX should be taken orally after eating, and with a full glass of water.
Starting Week DosageInstruct patients on how to titrate CHANTIX, beginning at a dose of 0.5 mg/day. Explain that one 0.5 mg tablet should be taken daily for the first three days, and that for the next four days, one 0.5 mg tablet should be taken in the morning and one 0.5 mg tablet should be taken in the evening.
Continuing Weeks DosageAdvise patients that, after the first seven days, the dose should be increased to one 1 mg tablet in the morning and one 1 mg tablet in the evening.
Dosage Adjustment For CHANTIX Or Other DrugsInform patients that nausea and insomnia are side effects of CHANTIX and are usually transient; however, advise patients that if they are persistently troubled by these symptoms, they should notify the prescribing physician so that a dose reduction can be considered.
Inform patients that some drugs may require dose adjustment after quitting smoking.
Counseling And SupportProvide patients with educational materials and necessary counseling to support an attempt at quitting smoking.
Neuropsychiatric Adverse EventsInform patients that some patients have experienced changes in mood (including depression and mania), psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, agitation, anxiety, and panic, as well as suicidal ideation and suicide when attempting to quit smoking while taking CHANTIX. Instruct patients to discontinue CHANTIX and contact a healthcare professional if they experience such symptoms.
History Of Psychiatric IllnessEncourage patients to reveal any history of psychiatric illness prior to initiating treatment.
Nicotine WithdrawalInform patients that quitting smoking, with or without CHANTIX, may be associated with nicotine withdrawal symptoms (including depression or agitation) or exacerbation of pre-existing psychiatric illness.
SeizuresEncourage patients to report any history of seizures or other factors that can lower seizure threshold. Instruct patients to discontinue CHANTIX and contact a healthcare provider immediately if they experience a seizure while on treatment.
Interaction With AlcoholAdvise patients to reduce the amount of alcohol they consume while taking CHANTIX until they know whether CHANTIX affects their tolerance for alcohol.
Driving Or Operating MachineryAdvise patients to use caution driving or operating machinery until they know how quitting smoking and/or varenicline may affect them.
Cardiovascular EventsPatients should be instructed to notify their healthcare providers of symptoms of new or worsening cardiovascular events and to seek immediate medical attention if they experience signs and symptoms of myocardial infarction or stroke.
SomnambulismPatients should be instructed to discontinue CHANTIX and notify their healthcare providers if they experience somnambulism.
AngioedemaInform patients that there have been reports of angioedema, with swelling of the face, mouth (lip, gum, tongue) and neck (larynx and pharynx) that can lead to life-threatening respiratory compromise. Instruct patients to discontinue CHANTIX and immediately seek medical care if they experience these symptoms.
Serious Skin ReactionsInform patients that serious skin reactions, such as Stevens-Johnson Syndrome and erythema multiforme, were reported by some patients taking CHANTIX. Advise patients to stop taking CHANTIX at the first sign of rash with mucosal lesions or skin reaction and contact a healthcare provider immediately.
Vivid, Unusual, Or Strange DreamsInform patients that they may experience vivid, unusual or strange dreams during treatment with CHANTIX.
Pregnancy And LactationPatients who are pregnant or breastfeeding or planning to become pregnant should be advised of: the risks of smoking to a pregnant mother and her developing baby, the potential risks of CHANTIX use during pregnancy and breastfeeding, and the benefits of smoking cessation with and without CHANTIX. Advise breastfeeding women to monitor the infant for seizures and vomiting.
This product's label may have been updated. For full prescribing information, please visit www.pfizer.com
Nonclinical Toxicology Carcinogenesis, Mutagenesis, Impairment Of Fertility CarcinogenesisLifetime carcinogenicity studies were performed in CD-1 mice and Sprague-Dawley rats. There was no evidence of a carcinogenic effect in mice administered varenicline by oral gavage for 2 years at doses up to 20 mg/kg/day (47 times the maximum recommended human daily (MRHD) exposure based on AUC). Rats were administered varenicline (1, 5, and 15 mg/kg/day) by oral gavage for 2 years. In male rats (n = 65 per sex per dose group), incidences of hibernoma (tumor of the brown fat) were increased at the mid dose (1 tumor, 5 mg/kg/day, 23 times the MRHD exposure based on AUC) and maximum dose (2 tumors, 15 mg/kg/day, 67 times the MRHD exposure based on AUC). The clinical relevance of this finding to humans has not been established. There was no evidence of carcinogenicity in female rats.
MutagenesisVarenicline was not genotoxic, with or without metabolic activation, in the following assays: Ames bacterial mutation assay; mammalian CHO/HGPRT assay; and tests for cytogenetic aberrations in vivo in rat bone marrow and in vitro in human lymphocytes.
Impairment Of FertilityThere was no evidence of impairment of fertility in either male or female Sprague-Dawley rats
administered varenicline succinate up to 15 mg/kg/day (67 and 36 times, respectively, the MRHD exposure based on AUC at 1 mg twice daily). Maternal toxicity, characterized by a decrease in body weight gain, was observed at 15 mg/kg/day. However, a decrease in fertility was noted in the offspring of pregnant rats who were administered varenicline succinate at an oral dose of 15 mg/kg/day. This decrease in fertility in the offspring of treated female rats was not evident at an oral dose of 3 mg/kg/day (9 times the MRHD exposure based on AUC at 1 mg twice daily).
Use In Specific Populations Pregnancy Risk SummaryAvailable human data on the use of CHANTIX in pregnant women are not sufficient to inform a drug associated risk. Smoking during pregnancy is associated with maternal, fetal, and neonatal risks. In animal studies, varenicline did not result in major malformations but caused decreased fetal weights in rabbits when dosed during organogenesis at exposures equivalent to 50 times the exposure at the maximum recommended human dose (MRHD). Additionally, administration of varenicline to pregnant rats during organogenesis through lactation produced developmental toxicity in offspring at maternal exposures equivalent to 36 times human exposure at the MRHD.
The estimated background risk of oral clefts is increased by approximately 30% in infants of women who smoke during pregnancy, compared to pregnant women who do not smoke. The background risk of other major birth defects and miscarriage for the indicated population are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Clinical ConsiderationsDisease-Associated Maternal And/Or Embryo/Fetal Risk
Smoking during pregnancy causes increased risks of orofacial clefts, premature rupture of membranes, placenta previa, placental abruption, ectopic pregnancy, fetal growth restriction and low birth weight, stillbirth, preterm delivery and shortened gestation, neonatal death, sudden infant death syndrome and reduction of lung function in infants. It is not known whether quitting smoking with CHANTIX during pregnancy reduces these risks.
DataAnimal Data
Pregnant rats and rabbits received varenicline succinate during organogenesis at oral doses up to 15 and 30 mg/kg/day, respectively. While no fetal structural abnormalities occurred in either species, maternal toxicity, characterized by reduced body weight gain, and reduced fetal weights occurred in rabbits at the highest dose (exposures 50 times the human exposure at the MRHD of 1 mg twice daily based on AUC). Fetal weight reduction did not occur in rabbits at exposures 23 times the human exposure at the MRHD based on AUC.
In a pre- and postnatal development study, pregnant rats received up to 15 mg/kg/day of oral varenicline succinate from organogenesis through lactation. Maternal toxicity, characterized by a decrease in body weight gain was observed at 15 mg/kg/day (36 times the human exposure at the MRHD based on AUC). However, decreased fertility and increased auditory startle response occurred in offspring at the highest maternal dose of 15 mg/kg/day.
Lactation Risk SummaryThere are no data on the presence of varenicline in human milk, the effects on the breastfed infant, or the effects on milk production. In animal studies varenicline was present in milk of lactating rats. However, due to species-specific differences in lactation physiology, animal data may not reliably predict drug levels in human milk. The lack of clinical data during lactation precludes a clear determination of the risk of CHANTIX to an infant during lactation; however the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CHANTIX and any potential adverse effects on the breastfed child from CHANTIX or from the underlying maternal condition.
Clinical ConsiderationsBecause there are no data on the presence of varenicline in human milk and the effects on the breastfed infant, breastfeeding women should monitor their infant for seizures and excessive vomiting, which are adverse reactions that have occurred in adults that may be clinically relevant in breastfeeding infants.
DataIn a pre- and postnatal development study, pregnant rats received up to 15 mg/kg/day of oral varenicline succinate through gestation and lactation Mean serum concentrations of varenicline in the nursing pups were 5–22% of maternal serum concentrations.
Pediatric UseSafety and effectiveness of CHANTIX in pediatric patients have not been established.
Geriatric UseA combined single- and multiple-dose pharmacokinetic study demonstrated that the pharmacokinetics of 1 mg varenicline given once daily or twice daily to 16 healthy elderly male and female smokers (aged 65–75 years) for 7 consecutive days was similar to that of younger subjects. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Varenicline is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
No dosage adjustment is recommended for elderly patients.
Renal ImpairmentVarenicline is substantially eliminated by renal glomerular filtration along with active tubular secretion. Dose reduction is not required in patients with mild to moderate renal impairment. For patients with severe renal impairment (estimated creatinine clearance <30 mL/min), and for patients with end-stage renal disease undergoing hemodialysis, dosage adjustment is needed.
Dosage (Posology) and method of administration
Usual Dosage For AdultsSmoking cessation therapies are more likely to succeed for patients who are motivated to stop smoking and who are provided additional advice and support. Provide patients with appropriate educational materials and counseling to support the quit attempt.
The patient should set a date to stop smoking. Begin CHANTIX dosing one week before this date. Alternatively, the patient can begin CHANTIX dosing and then quit smoking between days 8 and 35 of treatment.
CHANTIX should be taken orally after eating and with a full glass of water.
The recommended dose of CHANTIX is 1 mg twice daily following a 1-week titration as follows:
| Days 1 - 3: | 0.5 mg once daily |
| Days 4 - 7: | 0.5 mg twice daily |
| Day 8 - end of treatment: | 1 mg twice daily |
Patients should be treated with CHANTIX for 12 weeks. For patients who have successfully stopped smoking at the end of 12 weeks, an additional course of 12 weeks treatment with CHANTIX is recommended to further increase the likelihood of long-term abstinence.
For patients who are sure that they are not able or willing to quit abruptly, consider a gradual approach to quitting smoking with CHANTIX. Patients should begin CHANTIX dosing and reduce smoking by 50% from baseline within the first four weeks, by an additional 50% in the next four weeks, and continue reducing with the goal of reaching complete abstinence by 12 weeks. Continue CHANTIX treatment for an additional 12 weeks, for a total of 24 weeks of treatment. Encourage patients to attempt quitting sooner if they feel ready.
Patients who are motivated to quit, and who did not succeed in stopping smoking during prior CHANTIX therapy for reasons other than intolerability due to adverse events or who relapsed after treatment, should be encouraged to make another attempt with CHANTIX once factors contributing to the failed attempt have been identified and addressed.
Consider a temporary or permanent dose reduction in patients who cannot tolerate the adverse effects of CHANTIX.
Dosage In Special Populations Patients With Impaired Renal FunctionNo dosage adjustment is necessary for patients with mild to moderate renal impairment. For patients with severe renal impairment (estimated creatinine clearance less than 30 mL per min), the recommended starting dose of CHANTIX is 0.5 mg once daily. The dose may then be titrated as needed to a maximum dose of 0.5 mg twice daily. For patients with end-stage renal disease undergoing hemodialysis, a maximum dose of 0.5 mg once daily may be administered if tolerated.
Elderly And Patients With Impaired Hepatic FunctionNo dosage adjustment is necessary for patients with hepatic impairment. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
Interaction with other medicinal products and other forms of interaction
SIDE EFFECTSThe following serious adverse reactions were reported in postmarketing experience and are discussed in greater detail in other sections of the labeling:
- Neuropsychiatric Adverse Events including Suicidality
- Seizures
- Interaction with alcohol
- Accidental injury
- Cardiovascular events
- Somnambulism
- Angioedema and hypersensitivity reactions
- Serious skin reactions
In the placebo-controlled premarketing studies, the most common adverse events associated with CHANTIX (>5% and twice the rate seen in placebo-treated patients) were nausea, abnormal (vivid, unusual, or strange) dreams, constipation, flatulence, and vomiting.
The treatment discontinuation rate due to adverse events in patients dosed with 1 mg twice daily was 12% for CHANTIX, compared to 10% for placebo in studies of three months' treatment. In this group, the discontinuation rates that are higher than placebo for the most common adverse events in CHANTIX-treated patients were as follows: nausea (3% vs. 0.5% for placebo), insomnia (1.2% vs. 1.1% for placebo), and abnormal dreams (0.3% vs. 0.2% for placebo).
Smoking cessation, with or without treatment, is associated with nicotine withdrawal symptoms and has also been associated with the exacerbation of underlying psychiatric illness.
Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, the adverse reactions rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
During the premarketing development of CHANTIX, over 4500 subjects were exposed to CHANTIX, with over 450 treated for at least 24 weeks and approximately 100 for a year. Most study participants were treated for 12 weeks or less.
The most common adverse event associated with CHANTIX treatment is nausea, occurring in 30% of patients treated at the recommended dose, compared with 10% in patients taking a comparable placebo regimen.
Table 3 shows the adverse events for CHANTIX and placebo in the 12- week fixed dose premarketing studies with titration in the first week [Studies 2 (titrated arm only), 4, and 5]. Adverse events were categorized using the Medical Dictionary for Regulatory Activities (MedDRA, Version 7.1).
MedDRA High Level Group Terms (HLGT) reported in ≥ 5% of patients in the CHANTIX 1 mg twice daily dose group, and more commonly than in the placebo group, are listed, along with subordinate Preferred Terms (PT) reported in ≥ 1% of CHANTIX patients (and at least 0.5% more frequent than placebo). Closely related Preferred Terms such as 'Insomnia', 'Initial insomnia', 'Middle insomnia', 'Early morning awakening' were grouped, but individual patients reporting two or more grouped events are only counted once.
Table 3: Common Treatment Emergent AEs (%) in the
Fixed-Dose, Placebo- Controlled Studies (HLGTs ≥ 5% of patients in the 1
mg BID CHANTIX Group and more commonly than Placebo and PT ≥ 1% in the 1
mg BID CHANTIX Group, and 1 mg BID CHANTIX at least 0.5% more than Placebo)
| SYSTEM ORGAN CLASS High Level Group Term Preferred Term | CHANTIX 0.5 mg BID N=129 |
CHANTIX 1 mg BID N=821 |
Placebo N=805 |
| GASTROINTESTINAL (GI) | |||
| GI Signs and Symptoms | |||
| Nausea | 16 | 30 | 10 |
| Abdominal Pain * | 5 | 7 | 5 |
| Flatulence | 9 | 6 | 3 |
| Dyspepsia | 5 | 5 | 3 |
| Vomiting | 1 | 5 | 2 |
| GI Motility/Defecation Conditions | |||
| Constipation | 5 | 8 | 3 |
| Gastroesophageal reflux disease | 1 | 1 | 0 |
| Salivary Gland Conditions | |||
| Dry mouth | 4 | 6 | 4 |
| PSYCHIATRIC DISORDERS | |||
| Sleep Disorder/Disturbances | |||
| Insomnia† | 19 | 18 | 13 |
| Abnormal dreams | 9 | 13 | 5 |
| Sleep disorder | 2 | 5 | 3 |
| Nightmare | 2 | 1 | 0 |
| NERVOUS SYSTEM | |||
| Headaches | |||
| Headache | 19 | 15 | 13 |
| Neurological Disorders NEC | |||
| Dysgeusia | 8 | 5 | 4 |
| Somnolence | 3 | 3 | 2 |
| Lethargy | 2 | 1 | 0 |
| GENERAL DISORDERS | |||
| General Disorders NEC | |||
| Fatigue/Malaise/Asthenia | 4 | 7 | 6 |
| RESPIR/THORACIC/MEDIAST | |||
| Respiratory Disorders NEC | |||
| Rhinorrhea | 0 | 1 | 0 |
| Dyspnea | 2 | 1 | 1 |
| Upper Respiratory Tract Disorder | 7 | 5 | 4 |
| SKIN/SUBCUTANEOUS TISSUE | |||
| Epidermal and Dermal Conditions | |||
| Rash | 1 | 3 | 2 |
| Pruritis | 0 | 1 | 1 |
| METABOLISM & NUTRITION | |||
| Appetite/General mNutrition Disorders | |||
| Increased appetite | 4 | 3 | 2 |
| Decreased appetite/Anorexia | 1 | 2 | 1 |
| *Includes PTs Abdominal (pain, pain upper, pain lower,
discomfort, tenderness, distension) and Stomach discomfort †Includes PTs Insomnia/Initial insomnia/Middle insomnia/Early morning awakening |
|||
The overall pattern and frequency of adverse events during the longer-term premarketing trials was similar to those described in Table 3, though several of the most common events were reported by a greater proportion of patients with long-term use (e.g., nausea was reported in 40% of patients treated with CHANTIX 1 mg twice daily in a one year study, compared to 8% of placebo-treated patients).
Following is a list of treatment-emergent adverse events reported by patients treated with CHANTIX during all premarketing clinical trials and updated based on pooled data from 18 placebo-controlled preand post-marketing studies, including approximately 5,000 patients treated with varenicline. Adverse events were categorized using MedDRA, Version 16.0. The listing does not include those events already listed in the previous tables or elsewhere in labeling, those events for which a drug cause was remote, those events which were so general as to be uninformative, and those events reported only once which did not have a substantial probability of being acutely life-threatening.
Blood and Lymphatic System Disorders. Infrequent: anemia, lymphadenopathy. Rare: leukocytosis, splenomegaly, thrombocytopenia.
Cardiac Disorders. Infrequent: angina pectoris, myocardial infarction, palpitations, tachycardia. Rare: acute coronary syndrome, arrhythmia, atrial fibrillation, bradycardia, cardiac flutter, cor pulmonale, coronary artery disease, ventricular extrasystoles.
Ear and Labyrinth Disorders. Infrequent: tinnitus, vertigo. Rare: deafness, Meniere's disease.
Endocrine Disorders. Infrequent: thyroid gland disorders.
Eye Disorders. Infrequent: conjunctivitis, eye irritation, eye pain, vision blurred, visual impairment.
Rare: blindness transient, cataract subcapsular, dry eye, night blindness, ocular vascular disorder, photophobia, vitreous floaters.
Gastrointestinal Disorders. Frequent: diarrhea, toothache. Infrequent: dysphagia, eructation, gastritis, gastrointestinal hemorrhage, mouth ulceration. Rare: enterocolitis, esophagitis, gastric ulcer, intestinal obstruction, pancreatitis acute.
General Disorders and Administration Site Conditions. Frequent: chest pain. Infrequent: chest discomfort, chills, edema, influenza-like illness, pyrexia.
Hepatobiliary Disorders. Rare: gall bladder disorder.
Investigations. Frequent: liver function test abnormal, weight increased. Infrequent: electrocardiogram abnormal. Rare: muscle enzyme increased, urine analysis abnormal.
Metabolism and Nutrition Disorders. Infrequent: diabetes mellitus, hypoglycemia. Rare: hyperlipidemia, hypokalemia.
Musculoskeletal and Connective Tissue Disorders. Frequent: arthralgia, back pain, myalgia. Infrequent: arthritis, muscle cramp, musculoskeletal pain. Rare: myositis, osteoporosis.
Nervous System Disorders. Frequent: disturbance in attention, dizziness. Infrequent: amnesia, convulsion, migraine, parosmia, syncope, tremor. Rare: balance disorder, cerebrovascular accident, dysarthria, mental impairment, multiple sclerosis, VII th  nerve paralysis, nystagmus, psychomotor hyperactivity, psychomotor skills impaired, restless legs syndrome, sensory disturbance, transient ischemic attack, visual field defect.
Psychiatric Disorders. Infrequent: dissociation, libido decreased, mood swings, thinking abnormal. Rare: bradyphrenia, disorientation, euphoric mood.
Renal and Urinary Disorders. Infrequent: nocturia, pollakiuria, urine abnormality. Rare: nephrolithiasis, polyuria, renal failure acute, urethral syndrome, urinary retention.
Reproductive System and Breast Disorders. Frequent: menstrual disorder. Infrequent: erectile dysfunction. Rare: sexual dysfunction.
Respiratory, Thoracic and Mediastinal Disorders. Frequent: respiratory disorders. Infrequent: asthma, epistaxis, rhinitis allergic, upper respiratory tract inflammation. Rare: pleurisy, pulmonary embolism.
Skin and Subcutaneous Tissue Disorders. Infrequent: acne, dry skin, eczema, erythema, hyperhidrosis, urticaria. Rare: photosensitivity reaction, psoriasis.
Vascular Disorders. Infrequent: hot flush. Rare: thrombosis.
CHANTIX has also been studied in postmarketing trials including (1) a trial conducted in patients with chronic obstructive pulmonary disease (COPD), (2) a trial conducted in generally healthy patients (similar to those in the premarketing studies) in which they were allowed to select a quit date between days 8 and 35 of treatment (“alternative quit date instruction trial”), (3) a trial conducted in patients who did not succeed in stopping smoking during prior CHANTIX therapy, or who relapsed after treatment (“re-treatment trial”), (4) a trial conducted in patients with stable cardiovascular disease, (5) a trial conducted in patients with stable schizophrenia or schizoaffective disorder, (6) a trial conducted in patients with major depressive disorder, (7) a postmarketing neuropsychiatric safety outcome trial in patients without or with a history of psychiatric disorder, and (8) a trial in patients who were not able or willing to quit abruptly and who were instructed to quit gradually (“gradual approach to quitting smoking trial”).
Adverse events in the trial of patients with COPD, in the alternative quit date instruction trial, and in the gradual approach to quitting smoking trial were similar to those observed in premarketing studies. In the re-treatment trial, the profile of common adverse events was similar to that previously reported, but, in addition, varenicline-treated patients also commonly reported diarrhea (6% vs. 4% in placebo-treated patients), depressed mood disorders and disturbances (6% vs. 1%), and other mood disorders and disturbances (5% vs. 2%).
In the trial of patients with stable cardiovascular disease, more types and a greater number of cardiovascular events were reported compared to premarketing studies. Treatment-emergent (ontreatment or 30 days after treatment) cardiovascular events reported with a frequency ≥ 1% in either treatment group in this study were angina pectoris (3.7% and 2.0% for varenicline and placebo, respectively), chest pain (2.5% vs. 2.3%), peripheral edema (2.0% vs. 1.1%), hypertension (1.4% vs. 2.6%), and palpitations (0.6 % vs. 1.1%). Deaths and serious cardiovascular events occurring over the 52 weeks of the study (treatment emergent and non-treatment emergent) were adjudicated by a blinded, independent committee. The following treatment-emergent adjudicated events occurred with a frequency ≥ 1% in either treatment group: nonfatal MI (1.1% vs. 0.3% for varenicline and placebo, respectively), and hospitalization for angina pectoris (0.6% vs. 1.1%). During non-treatment follow-up to 52 weeks, the adjudicated events included need for coronary revascularization (2.0% vs. 0.6%), hospitalization for angina pectoris (1.7% vs. 1.1%), and new diagnosis of peripheral vascular disease (PVD) or admission for a PVD procedure (1.4% vs. 0.6%). Some of the patients requiring coronary revascularization underwent the procedure as part of management of nonfatal MI and hospitalization for angina. Cardiovascular death occurred in 0.3% of patients in the varenicline arm and 0.6% of patients in the placebo arm over the course of the 52-week study.
In the trial of patients with stable schizophrenia or schizoaffective disorder, 128 smokers on antipsychotic medication were randomized 2:1 to varenicline (1 mg twice daily) or placebo for 12 weeks with 12-week non-drug follow-up. The most common adverse events in patients taking varenicline were nausea (24% vs. 14.0% on placebo), headache (11% vs. 19% on placebo) and vomiting (11% vs. 9% on placebo). Among reported neuropsychiatric adverse events, insomnia was the only event that occurred in either treatment group in ≥5% of subjects at a rate higher in the varenicline group than in placebo (10% vs. 5%). These common and neuropsychiatric adverse events occurred on treatment or within 30 days after the last dose of study drug. There was no consistent worsening of schizophrenia in either treatment group as measured by the Positive and Negative Syndrome Scale. There were no overall changes in extra-pyramidal signs, as measured by the Simpson-Angus Rating Scale. The Columbia-Suicide Severity Rating Scale was administered at baseline and at clinic visits during the treatment and non-treatment follow-up phases. Over half of the patients had a lifetime history of suicidal behavior and/or ideation (62% on varenicline vs. 51% on placebo), but at baseline, no patients in the varenicline group reported suicidal behavior and/or ideation vs. one patient in the placebo group (2%). Suicidal behavior and/or ideation were reported in 11% of the varenicline-treated and 9% of the placebo-treated patients during the treatment phase. During the post-treatment phase, suicidal behavior and/or ideation were reported in 11% of patients in the varenicline group and 5% of patients in the placebo group. Many of the patients reporting suicidal behavior and ideation in the follow-up phase had not reported such experiences in the treatment phase. However, no new suicidal ideation or behavior emerged in either treatment group shortly (within one week) after treatment discontinuation (a phenomenon noted in postmarketing reporting). There were no completed suicides. There was one suicide attempt in a varenicline-treated patient. The limited data available from this single smoking cessation study are not sufficient to allow conclusions to be drawn.
In the trial of patients with major depressive disorder, the most common adverse events (≥ 10%) in subjects taking varenicline were nausea (27% vs. 10% on placebo), headache (17 vs. 11%), abnormal dreams (11% vs. 8%), insomnia (11% vs. 5%) and irritability (11% vs. 8%). Additionally, the following psychiatric AEs were reported in ≥ 2% of patients in either treatment group (varenicline or placebo, respectively): anxiety (7% vs. 9%), agitation (7% vs. 4%), depressed mood disorders and disturbances (11% vs. 9%), tension (4% vs. 3%), hostility (2% vs. 0.4%) and restlessness (2% vs. 2%). Patients treated with varenicline were more likely than patients treated with placebo to report one of various events related to hostility and aggression (3% vs. 1%). Psychiatric scales showed no differences between the varenicline and placebo groups and no overall worsening of depression during the study in either treatment group. The percentage of subjects with suicidal ideation and/or behavior was similar between the varenicline and placebo groups during treatment (6% and 8%, respectively) and the nontreatment follow-up (6% and 6%, respectively). There was one event of intentional self-injury/possible suicide attempt during treatment (Day 73) in a subject in the placebo group. Suicide could not be ruled out in one subject who died by an overdose of illicit drugs 76 days after last dose of study drug in the varenicline group.
In the trial of patients without or with a history of psychiatric disorder, the most common adverse events in subjects treated with varenicline were similar to those observed in premarketing studies. Adverse events reported in ≥ 10% of subjects treated with varenicline in the entire study population were nausea (25% vs. 7% on placebo) and headache (12% vs. 10% on placebo). Additionally, the following psychiatric adverse events were reported in ≥ 2% of patients in either treatment group (varenicline vs. placebo) by cohort. For the non-psychiatric cohort, these adverse events were abnormal dreams (8% vs. 4%), agitation (3% vs. 3%), anxiety (5% vs. 6%), depressed mood (3% vs. 3%), insomnia (10% vs. 7%), irritability (3% vs. 4%), sleep disorder (3% vs. 2%). For the psychiatric cohort, these adverse events were abnormal dreams (12% vs. 5%), agitation (5% vs. 4%), anxiety (8% vs. 6%), depressed mood (5% vs. 5%), depression (5% vs. 5%), insomnia (9% vs. 7%), irritability (5% vs. 7%), nervousness (2% vs. 3%), sleep disorder (3% vs. 2%).
Postmarketing ExperienceThe following adverse events have been reported during post-approval use of CHANTIX. Because these events are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
There have been reports of depression, mania, psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, anxiety, and panic, as well as suicidal ideation, suicide attempt, and completed suicide in patients attempting to quit smoking while taking CHANTIX.
There have been postmarketing reports of new or worsening seizures in patients treated with CHANTIX.
There have been postmarketing reports of patients experiencing increased intoxicating effects of alcohol while taking CHANTIX. Some reported neuropsychiatric events, including unusual and sometimes aggressive behavior.
There have been reports of hypersensitivity reactions, including angioedema.
There have also been reports of serious skin reactions, including Stevens-Johnson Syndrome and erythema multiforme, in patients taking CHANTIX. There have been reports of myocardial infarction (MI) and cerebrovascular accident (CVA) including ischemic and hemorrhagic events in patients taking CHANTIX. In the majority of the reported cases, patients had pre-existing cardiovascular disease and/or other risk factors. Although smoking is a risk factor for MI and CVA, based on temporal relationship between medication use and events, a contributory role of varenicline cannot be ruled out.
There have been reports of hyperglycemia in patients following initiation of CHANTIX. There have been reports of somnambulism, some resulting in harmful behavior to self, others, or property in patients treated with CHANTIX.
DRUG INTERACTIONSBased on varenicline characteristics and clinical experience to date, CHANTIX has no clinically meaningful pharmacokinetic drug interactions.
Use With Other Drugs For Smoking CessationSafety and efficacy of CHANTIX in combination with other smoking cessation therapies have not been studied.
BupropionVarenicline (1 mg twice daily) did not alter the steady-state pharmacokinetics of bupropion (150 mg twice daily) in 46 smokers. The safety of the combination of bupropion and varenicline has not been established.
Nicotine Replacement Therapy (NRT)Although co-administration of varenicline (1 mg twice daily) and transdermal nicotine (21 mg/day) for up to 12 days did not affect nicotine pharmacokinetics, the incidence of nausea, headache, vomiting, dizziness, dyspepsia, and fatigue was greater for the combination than for NRT alone. In this study, eight of twenty-two (36%) patients treated with the combination of varenicline and NRT prematurely discontinued treatment due to adverse events, compared to 1 of 17 (6%) of patients treated with NRT and placebo.
Effect Of Smoking Cessation On Other DrugsPhysiological changes resulting from smoking cessation, with or without treatment with CHANTIX, may alter the pharmacokinetics or pharmacodynamics of certain drugs (e.g., theophylline, warfarin, insulin) for which dosage adjustment may be necessary.
Drug Abuse And Dependence Controlled SubstanceVarenicline is not a controlled substance.
Dependence HumansFewer than 1 out of 1,000 patients reported euphoria in clinical trials with CHANTIX. At higher doses (greater than 2 mg), CHANTIX produced more frequent reports of gastrointestinal disturbances such as nausea and vomiting. There is no evidence of dose-escalation to maintain therapeutic effects in clinical studies, which suggests that tolerance does not develop. Abrupt discontinuation of CHANTIX was associated with an increase in irritability and sleep disturbances in up to 3% of patients. This suggests that, in some patients, varenicline may produce mild physical dependence which is not associated with addiction.
In a human laboratory abuse liability study, a single oral dose of 1 mg varenicline did not produce any significant positive or negative subjective responses in smokers. In non-smokers, 1 mg varenicline produced an increase in some positive subjective effects, but this was accompanied by an increase in negative adverse effects, especially nausea. A single oral dose of 3 mg varenicline uniformly produced unpleasant subjective responses in both smokers and non-smokers.
AnimalsStudies in rodents have shown that varenicline produces behavioral responses similar to those produced by nicotine. In rats trained to discriminate nicotine from saline, varenicline produced full generalization to the nicotine cue. In self-administration studies, the degree to which varenicline substitutes for nicotine is dependent upon the requirement of the task. Rats trained to self-administer nicotine under easy conditions continued to self-administer varenicline to a degree comparable to that of nicotine; however in a more demanding task, rats self-administered varenicline to a lesser extent than nicotine. Varenicline pretreatment also reduced nicotine self-administration.